
Abstract
The gut microbiota plays a critical role in host health, yet remains poorly studied in wild species. Polar bears (Ursus maritimus), key indicators of Arctic ecosystem health and environmental change, are currently affected by rapid shifts in habitat that may alter gut homeostasis. Declining sea ice has led to a divide in the southern Beaufort Sea polar bear subpopulation such that an increasing proportion of individuals now inhabit onshore coastal regions during the open-water period (‘onshore bears’) while others continue to exhibit their typical behaviour of remaining on the ice (‘offshore bears’). We propose that bears that have altered their habitat selection in response to climate change will exhibit a distinct gut microbiota diversity and composition, which may ultimately have important consequences for their health. Here, we perform the first assessment of abundance and diversity in the faecal microbiota of wild polar bears using 16S rRNA Illumina technology. We find that bacterial diversity is significantly higher in onshore bears compared to offshore bears. The most enriched OTU abundance in onshore bears belonged to the phylum Proteobacteria, while the most depleted OTU abundance within onshore bears was seen in the phylum Firmicutes. We conclude that climate-driven changes in polar bear land use are associated with distinct microbial communities. In doing so, we present the first case of global change mediated alterations in the gut microbiota of a free-roaming wild animal.
Similar content being viewed by others


Introduction
As an apex predator with vulnerable conservation status [1], the polar bear (Ursus maritimus) is widely acknowledged as a key indicator of Arctic ecosystem health [2], a model species for studying the effects of climatic and other anthropogenic stressors in the Arctic [3,4,5], and a flagship for environmental change [6]. As one of the most ice dependent Arctic marine mammals [7], polar bears require sea ice for long-distance movements, mating and accessing prey [8]. One subpopulation of polar bear, the southern Beaufort Sea subpopulation, is exhibiting a distinct behavioural response to climate-driven changes in sea ice conditions. Historically, these polar bears remained year-round on the sea ice (hereafter referred to as ‘offshore bears’), taking advantage of the biologically productive continental shelf [9]. Since the 2000s, however, substantial declines in the spatial and temporal availability of sea ice in summer and fall [10, 11], extending well beyond the continental shelf, have driven a divide in polar bear behaviour whereby some continue to select the retreating ice habitat (‘offshore bears’) while others instead adopt a novel behaviour and move to coastal onshore habitat during the reduced ice period (‘onshore bears’) [12]. The entire subpopulation uses the sea ice during the remainder of the year. Onshore bears have been associated with a range of dietary items that offshore bears are unable to access, notably ‘bone piles’, the remains of locally harvested bowhead whales (Balaena mysticetus), along with the carcasses of fish, birds and caribou (Rangifer tarandus) [13]. Conversely, offshore bears primarily consume a traditional diet of ringed seal (Pusa hispida), bearded seal (Erignathus barbatus) and occasionally beluga whale (Delphinapterus leucas) [13], which are inaccessible to onshore bears.
Changes in trophic interactions alter the exposure of polar bears to contaminants and novel parasites [14, 15]. For example, ringed seals (available only to offshore bears) are considered to occupy a high trophic position and so typically bioaccumulate higher levels of contaminants than species lower in the trophic chain such as the filter feeders (i.e. bowhead whales) and herbivores (i.e. caribou) [16,17,18], which are available only to onshore bears. In addition, bone piles, foraged on by onshore bears, are utilised as a food resource by other terrestrial species [13, 19] and lie within comparatively close range of human settlements, such as Kaktovik (70.13°N, 143.62°W) and Deadhorse (70.20°N, 148.46°W). Thus, onshore bears are potentially exposed to (and therefore at greater risk of infection from) novel parasites carried by terrestrial species, including humans and their domestic pets. For example, Atwood et al. (2017) [5] found that southern Beaufort Sea polar bears exhibiting onshore behaviour have a greater risk of exposure to Toxoplasma gondii and lower exposure to certain contaminants than offshore bears. Thus, onshore bears are exposed to different biotic stressors compared to offshore bears [5, 20], which have the potential to drive variation in the gut microbiota. In humans and mice, for example, helminth infection is associated with significant differences in the community composition of gut bacterial communities [21,22,23], while contaminants such as herbicides and pesticides have been shown to inhibit the growth of a variety of beneficial gut bacteria [24] and even cause dysbiosis [25].
The gut microbiota, a diverse community of bacteria that resides within the gastrointestinal tract, has a long co-evolutionary association with its host [26], carrying out vital nutritional and physiological roles [26,27,28]. In effect, the regular intestinal development and function of an individual is attributed to an array of specific bacterial groups or species, the composition and diversity of which are a function of complex interactions between host and environment [29]. Despite the importance of the gut microbiota to health, little is understood of the composition or community structure of the gut microbiota of wild fauna [30]. In brown bears (U. arctos) however, we know a distinct gut microbiota profile is associated with active bears compared to those in hibernation phase—this specific community of bacteria is thought to play a role in promoting adiposity while still maintaining normal gut metabolism [31]. A paucity of knowledge on wild microbiota is particularly concerning considering that in the face of rapid climate change tight host-gut microbiota associations could quickly become decoupled, negating millions of years of co-evolutionary adaptation [26], and yet this too remains poorly understood.
A number of studies provide support for an association between host microbial communities and environmental fluctuations. Cold acclimated laboratory mice, for example, harbour a dramatically different gut microbiota composition to those raised at higher temperatures [32], while experimentally induced temperature increases of 2–3 °C cause a 34% loss of microbiota diversity in the common lizard (Zootoca vivipara) [33]. Outside a laboratory setting, variations in weather events have been linked to the increased occurrence of gastrointestinal illness in residents of Nunatsiavut, Canada [34]. To the best of our knowledge, however, no study has demonstrated a climate change mediated alteration in the gut microbiota of free-roaming wildlife.
The gut microbiota has been examined once before in wild polar bears, specifically those from the Svalbard archipelago belonging to the Barents Sea subpopulation [35]. The authors found a low bacterial diversity, dissimilar to that reported in other Arctic carnivores [36] and wild ursids [31, 37, 38], possibly attributed to the methodologies employed (having used 16S rRNA clone libraries as opposed to next generation sequencing techniques) and small sample size [35, 39]. Here we use high-throughput sequencing techniques to conduct the first detailed investigation of the gut microbiota composition of a large sample (n = 112) of wild southern Beaufort Sea polar bears and to establish the diversity, abundance, and composition of gut bacteria associated with on- and offshore bears. In doing so, we are able to evaluate the effect of a climate driven change in habitat use on microbial composition. Reflecting methods widely used in other gut microbiota studies [40], we use faeces as a proxy of gut microbiota, herein referred to as the faecal microbiota.
Materials and methods
Polar bear capture and sampling
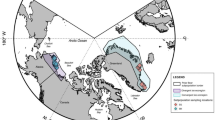
Polar bears were captured under the United States Geological Survey (USGS) Polar Bear Research Program (Marine Mammal Permit MA690038 to T.C.A.) in an area ranging approximately from Utqiagvik, Alaska (156°W) in the west to Demarcation Point (140°W) at the US-Canada border in the east, and extending from the shoreline to ~135 km north on sea ice (with the exception of one individual; Fig. 1). In the spring and fall of 2008 and 2009, and the spring of 2010 and 2013, polar bears were encountered via helicopter and immobilised with a remote injection of zolazepam-tiletamine (Telazol®, Fort Dodge Animal Health, Fort Dodge, Iowa, USA, and Warner-Lambert Co., Groton, Connecticut, USA). A single faecal sample was collected directly from the rectum of each polar bear using a sterile latex glove and immediately transferred to a sterile Whirl-pak bag (Nasco, Fort Atkinson, Wisconsin, USA) for storage. In total, samples were taken from 112 individuals, including 89 adults and 23 subadults, (51 males and 61 females). All samples were stored at −20 °C for the duration of the field season (~5 weeks) before being stored at −80 °C at the US Geological Survey, Alaska Science Center (Anchorage, Alaska, USA), and subsequently shipped on dry ice to the Fondazione Edmund Mach, Italy (CITES permit IT/IM/2015/MCE/01862 to S.W.).

Map of study area showing the sampling locations of 112 southern Beaufort Sea polar bears along the north coast of Alaska. Inset map shows the location of the study area, highlighting that one sample originates from a more northerly location that the others
Age of subadults and adults was estimated by extracting and analysing the cementum annuli of a vestigial premolar tooth [41]. In total, 85 of the 112 bears were known to be either onshore or offshore (onshore n = 46; offshore n = 39; Supplementary Table 1). Individuals were categorised as either ‘onshore bears’ or ‘offshore bears’ as described in [5]. In brief, location data collected from satellite collars were used to identify adult females that used land (‘onshore’) or sea ice (‘offshore’) in summer and fall [42]. We classified both male and female individuals as onshore bears if they were detected (via genetic identification and cross-referencing with our database of known bears) at hair-snags erected in the fall around bowhead whale bone piles and from biopsy-darting during fall coastal surveys from 2010 to 2013. An individual was classified as onshore or offshore if spatial or genetic data suggested that the individual was onshore or offshore in summer and/or in the year of capture (for fall-captured bears) or immediately prior to capture (for spring-captured bears). Body condition for each polar bear was estimated using a ‘Body Condition Index’ metric [43] and was classified as either above or below the mean body condition for our sample set. Year and season of capture was also recorded.
Extraction of bacterial DNA
All faecal matter was collected from inside each sample glove using a sterile cotton swab (APTACA sterile transport swabs, Brescia, Italy). The swab was subsequently vortexed for 10 min in 1 ml phosphate-buffered saline solution (PBS) and pelleted by centrifugation at 16,000 g for 12 min. Lysis buffer, 80 µl, (200 mM NaCl, 100 mM Tris, 20 mM EDTA, 20 mg/ml Lysozyme, pH 8.0); 5 mm stainless steel beads (Qiagen) were added to each sample before a three-minute homogenisation step at 30 Hz using a Mixer Mill MM200 (Retsch GmbH, Haan, Germany). Samples were then shaken at 37 °C for 40 min Grant-Bio PCMT Thermoshaker (500 rpm). Microbial DNA was extracted using the QIAamp® DNA Mini Kits (QIAGEN©, Milan, Italy), following the manufacturer’s Buccal Swab Spin Protocol for cotton swabs (QIAamp® DNA Mini and Blood Mini Handbook), but starting from step 2 (addition of Proteinase K).
16S rRNA gene amplification and sequencing
Using the bacteria-specific primer set 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805Rmod (5′-GACTACNVGGGTWTCTAATCC-3′) (based on Klindworth et al. [44] with degenerate bases) with overhanging Illumina adaptors, a ~460 base pair (bp) fragment of the 16S rRNA gene (variable region V3-V4) [45] was amplified using a GeneAmp PCR System 9700 (Thermo Fisher Scientific) and the following steps: 94 °C for 5 min (one cycle), 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s (30 cycles), 72 °C for 5 min (1 cycle). The PCR products were visualised on a 1.5% agarose gel and purified using Agencourt AMPure XP SPRI beads (Beckman Coulter, Brea, CA, USA) following manufacturer’s instructions. Subsequently, Illumina® Nextera XT indices and sequencing adaptors (Illumina®) were incorporated using seven cycles of PCR (16S Metagenomic Sequencing Library Preparation, Illumina®). The final libraries were quantified using the Quant-IT PicoGreen dsDNA assay kit (Thermo Fisher Scientific) by the Synergy2 microplate reader (Biotek), pooled in equimolar concentration before sequencing on an lllumina® MiSeq (2 × 300 bp reads) at the Next Generation Sequencing Platform, Fondazione Edmund Mach in collaboration with the Core Facility, CIBIO, University of Trento, Italy. All samples were sequenced in one Illumina MiSeq Standard Flow Cell targeting a depth of 20,000 reads per sample.
Bioinformatic processing of 16S data
Reads were processed with MICCA v1.5.0 [46]. In brief, paired-end reads were merged, and pairs diverging by more than 8 bp or overlapping by <100 bp were discarded. PCR amplification primers were trimmed (sequences not containing both PCR primer sequences were discarded). Finally, sequences were quality filtered at 0.5% Expected Error (EE); those displaying greater than 0.5% EE were discarded along with those shorter than 400 bp or containing unknown base calls (N). Using the VSEARCH cluster_smallmem algorithm [47], OTUs were created de novo by clustering sequences with 97% sequence identity, discarding chimeric sequences. Taxonomic assignments of representative sequences from each OTU were performed using the RDP Classifier v2.12 in conjunction with RDP 16S rRNA training set 15 [48]. OTU sequences were aligned and phylogenetic analysis was performed using Nearest Alignment Space Termination (NAST) and a phylogeny reconstructed using FastTree [49], both via MICCA [46]. The raw sequencing data can be found at the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) [Accession number: PRJNA542176].
Statistical analyses
Following initial processing, singletons were removed and all samples with fewer than 5000 reads were removed using the R package ‘phyloseq’ [50], leaving a total of 511,952 reads across 112 samples. The data were rarefied to an equal depth within 90% of the minimum observed sample size (specifically 4571 reads per sample). Generalized Linear Models (GLMs) with a Gamma error function were used to investigate whether metadata (onshore/offshore, age class, sex, body condition, year of capture and season of capture) were associated with alpha diversity of the faecal microbiota (Shannon, Inverse Simpson and Faith’s Phylogenetic Diversity Indices). For Shannon and Faith’s Phylogenetic Diversity measures, an identity link function was used, while a log link function was used when analysing an Inverse Simpson measure of diversity. All multivariate analyses on faecal microbiota structure according to host metadata (on-/offshore, age class, sex, body condition, year of capture and season) were assessed using PERMANOVA, based on Bray-Curtis dissimilarity and weighted UniFrac indices, using the ‘adonis’ function in the R package ‘vegan’ [51]. An important assumption for PERMANOVA is homogenous dispersion of data among groups; for this reason, the ‘betadisper’ function in ‘vegan’ was implemented to investigate the homogeneity of data. Data rows containing missing values (NAs) were removed from the dataset prior to conducting the PERMANOVA to ensure matrices were even between variables. To determine the differential abundance of OTUs between on- and offshore bears, sex and season were examined using the R package ‘DESeq2’ [52]. To assess whether the microbiota profiles of polar bears is related to their geographic distribution, a GPS-based pairwise distance matrix was constructed using the R package ‘geosphere’ [53] and compared to a PCoA matrix (using both Bray-Curtis and weighted UniFrac) via a Mantel Test. All analyses were carried out using R statistical software package, version 3.2.0 [54]. Data were visualised using the R packages ‘ggplot2’ [55] and ‘metacoder’ [56].
Results
Faecal microbiota composition
The faecal microbiota of all 112 bears was composed of 1221 operational taxonomic units (OTUs) encompassing 25 bacterial phyla, with prevalence and abundance of specific phyla differing among individuals (Fig. 2a). Across the population, the most abundant phyla (which composed 91% of the total reads and were present in all individuals) were Firmicutes (45%), Proteobacteria (25%) and Actinobacteria (21%), making up the core microbiota. All other phyla represented <9% of reads each (Fig. 2a), and their prevalence among samples varied between 97% (Bacteroidetes) and 1% (Armatimonadetes, Deferribacteres, Lentisphaerae and Synergistetes). From the total number of reads obtained for the most dominant phylum (Firmicutes), 70% belonged to the class Clostridia, and 99% of those were from the order Clostridiales. The dominant orders for the remaining top bacterial phyla were Enterobacteriales (phyla: Proteobacteria) and Actinomycetales (phyla: Actinobacteria) (Fig. 2b).

a Stacked bar chart of the relative abundance of 25 bacterial phyla in the faecal microbiota of 112 southern Beaufort Sea polar bears. Phyla in the legend are listed in order of decreasing abundance. b Inset is a metacoder heatmap plotted to order level: each node moving from the centre outwards represents a different taxonomic rank, whereby kingdom is the centre and nodes representing order appear on the outer edges. The map is weighted and coloured by read abundance
Onshore versus offshore microbiota
Using the subset of bears for which we had on- and offshore information (n = 85), we found alpha diversity was significantly higher in on- (n = 46) compared to offshore (n = 39) bears, for Shannon (adjusted R2 = 0.06, F1,83 = 6.32, P = 0.014; Fig. 3a and Supplementary Table 2) and Inverse Simpson (adjusted R2 = 0.07, F1,83 = 6.09, P = 0.016; Fig. 3b and Supplementary Table 2) indices but not for Faith’s Phylogenetic Diversity index (Supplementary Table 3). Beta diversity did not differ between on- and offshore bears when using Bray-Curtis (Supplementary Fig. 1) but differed significantly between on- and offshore bears when using a weighted UniFrac metric (adjusted R2 = 0.03, F1,80 = 2.53, P = 0.029; Supplementary Fig. 2). Data dispersion did not significantly differ between on- and offshore bears (P = 0.740).

Violin plots of alpha diversity within the faecal microbiota of 85 southern Beaufort Sea polar bears for which ‘onshore/offshore’ land use is known (see text for definitions): a Shannon diversity index. b Inverse Simpson diversity index. Violin plots combine a box plot with a density plot, and as such the width of each plot corresponds to the distribution of the data
The faecal microbiota of onshore bears consisted of 858 OTUs (19 bacterial phyla; 37 classes) compared to 635 OTUs (21 phyla; 35 classes) for offshore bears, of which 386 were shared between on- and offshore polar bears (Fig. 4). Of the total number of OTUs found 472 were unique to onshore bears, and a smaller number of OTUs (n = 249) were unique to offshore bears. Eleven OTUs (10 Firmicutes; 1 Proteobacteria) were significantly enriched and 6 OTUs (3 Bacteroidetes; 2 Firmicutes; 1 Proteobacteria) were significantly reduced in onshore bears (Fig. 5; Supplementary Table 4). The majority (73%; n = 8) of OTUs that were enriched in onshore bears belonged to the order Clostridiales (Phylum: Firmicutes), although family level assignment varied across OTUs (Fig. 5 and Supplementary Table 4). OTUs that were significantly decreased in on- compared to offshore bears varied in taxonomic assignment across taxonomic ranks (Supplementary Table 4). The most enriched OTU abundance in onshore bears belonged to the family Moraxellaceae (Phylum: Proteobacteria), with a 6.78 log2 fold change in abundance (P < 0.001), while the most depleted OTU abundance within onshore bears was seen in Clostridiaceae 1 (Phylum: Firmicutes) with a −8.04 log2 fold change in abundance (P < 0.001; Supplementary Table 4).

Log abundance of OTUs in the faecal microbiota of ‘onshore’ and ‘offshore’ bears, by bacterial Class. Inset shows shared number of OTUs by onshore (green) and offshore (blue) bears

Differential OTU abundance of onshore compared to offshore bears from DESeq2 analysis, plotted with individual OTU number and associated family assignment
The gut microbiota composition of individuals was not associated with their geographic proximity to one another (P = 0.56 and P = 0.17; Mantel Test using Bray-Curtis and weighted Unifrac respectively).
Ecological factors and the microbiota
When using Faith’s Phylogenetic Diversity Index, alpha diversity was significantly higher in females compared to males (adjusted R2 = 0.30, F2,109 = 25.18, P = 0.017), as well as in fall compared to spring captures (adjusted R2 = 0.30, F2,109 = 25.18, P < 0.001). However, alpha diversity did not differ with sex, season of capture, body condition, year or age class when using either a Shannon or Inverse Simpson index of diversity and no significant difference in alpha diversity was seen with body condition, year or age class when using Faith’s Phylogenetic Diversity. Beta diversity differed significantly with sex (Bray-Curtis; P = 0.001; weighted UniFrac P = 0.006) although data dispersion was seen to be significantly different between males and females (P = 0.018) and so the PERMANOVA should be interpreted with caution. Beta diversity also differed significantly with season when using Bray-Curtis (P = 0.005) but not weighted UniFrac (P = 0.184), where beta dispersion was P = 0.113. No differences in beta diversity were seen with year, age class or body condition when using either Bray-Curtis or a weighted UniFrac metric. When investigating the differential abundance of OTUs with sex, DESeq analysis showed that 66 OTUs were significantly different between males and females; 9 OTUs were significantly increased in males compared to females (the largest increase, of 5.40 log fold change, belonging to the family Clostridiales Incertae Sedis XI, phylum: Firmicutes) and 57 OTUs were significantly decreased (the largest decrease, of −10.04 log fold change, being seen in the family Flavobacteriaceae, phylum: Bacteroidetes). For season of capture, DESeq analysis revealed that 15 OTUs were significantly different between fall and spring captures; 2 OTUs were increased in spring compared to fall captures (the largest increase, of 3.01 log fold change, belonging to the family Veillonellaceae, phylum: Firmicutes) and 13 OTUs were significantly decreased (the largest decrease, of −7.50 log fold change, being seen in the family Peptostreptococcaceae, phylum: Firmicutes).
Discussion
Investigating factors which may influence the gut microbiota in a sentinel species experiencing rapid environmental change may improve our understanding of the role of the gut microbiota in wildlife health and conservation. Here we have shown that for the southern Beaufort Sea subpopulation of polar bears alpha diversity and bacterial composition are significantly different in the gut of onshore bears compared to those that remain on the sea ice year-round. As such, our study shows for the first time, that global change driven alterations in habitat use are associated with changes in the gut microbial composition and diversity of a free-ranging species.
We detected 25 bacterial phyla, as opposed to just the one (Firmicutes) previously found by Glad et al. [35] in wild Barents Sea polar bears. This diversity closely mirrors that seen in other studies utilising next generation sequencing methods to investigate the gut microbiota of ursids; for example, 24 bacterial phyla were detected in wild brown bears [31]. The most abundant phyla in polar bear faeces (Firmicutes, Proteobacteria and Actinobacteria), coincided with those of the core mammalian gut microbiota [26], including that of Asiatic black bears (Ursus thibetanus) [38]. Our finding that Firmicutes constituted the majority of OTUs is noteworthy in that increased Firmicutes in genetically obese mice and humans suggests that this phylum plays an important role in promoting adiposity or energy resorption [57], although conflicting studies show no link between Firmicutes levels and obesity/high-fat intake [58]. Interestingly, brown bears gaining weight for hibernation during summer months show simultaneously elevated levels of Firmicutes in the gut [31], implying this phylum may also play a role in synthesising high energy inputs in large carnivores. More specifically, we show that 70% of reads assigned to the phylum Firmicutes belonged to the class Clostridia, and subsequently 99% were from the order Clostridiales—an outcome that coincides with the results of Glad et al. [35], who showed all except one of the gene clones generated within their study were affiliated with the order Clostridiales. In a study using both wild type and laboratory mice, Hilderbrant et al. [59] showed that levels of Clostridiales greatly increases after prolonged durations of time feeding on a high-fat diet.
Within this study we found that alpha diversity of bacterial OTUs was significantly higher in the faecal microbiota of onshore compared to offshore bears when using a Shannon or Inverse Simpson measure, but no association was found between alpha diversity and host metadata (age class, sex, body condition, year or season of capture) when using these indices. Much microbiota work focusing on humans has found sex and age influences microbiota dynamics [60,61,62]. Although the majority of microbiota research has focused on humans, microbial studies of wild animals are increasing [30] and in some cases wild animals have been shown to follow similar trait-related stratification in microbiota. For example, the presence/absence of specific bacterial taxa were seen to correlate with specific age classes within the gut microbiota of wild ring-tailed lemurs (Lemur catta) [63]. Similarly, sex-specific differences in bacterial diversity have been found in, for example, wild rufous mouse lemurs (Microcebus rufus), whereby females demonstrated higher bacterial diversity compared to their male counterparts [64]. Further to this, season of capture has been seen to influence the gut microbiota composition. Sommer et al. [31], for example, demonstrated that gut microbial composition of free-roaming brown bears is seasonally altered between summer and winter. This change in bacterial composition is thought to, in part, be influenced by extreme dietary shifts within brown bears between active and hibernation phase [30]. We also see this seasonal shift in gut microbial composition in other wild animal models such as wild wood mice (Apodemus sylvaticus) [65], wild black howler monkey (Alouatta pigra) [66] and the giant panda (Ailuropoda melanoleuca) [37], probably also attributable to season-driven shifts in diet. None of these factors, however, were found to influence the gut microbiota composition of the polar bears sampled within this study when using a Shannon and Inverse Simpson index of diversity. However, when using Faith’s Phylogenetic Diversity (i.e. a metric that characterises only the relatedness or distinctness of species and works under the assumption that different species make unequal contributions to diversity [67]) we see a significant difference in diversity with sex and season only, whereby females had a higher bacterial diversity than males, and fall captures had a higher bacterial diversity than spring captures. Faith’s phylogenetic diversity index does not incorporate the relative abundances of taxa within communities, but rather calculates phylogenetic diversity based on the presence or absence of species [68, 69]. Our results therefore imply that for sex and season, there was no difference in alpha diversity when considering the richness and evenness of species, but that there may be a number of species with deep and/or distinct branching that are making an unequal contribution to the diversity of those communities.
We posit that the differences in gut microbiota composition between on- and offshore bears is most likely driven by environmental factors, such as diet, contaminants and parasites which are known to differ between the two groups [5, 12, 70, 71] although this hypothesis is yet to be tested. Diet, as one of the biggest drivers in gut microbial changes [72,73,74], likely plays the largest role in the observed differences in bacterial diversity. Historically, southern Beaufort Sea polar bears remained offshore hunting ringed seal (Pusa hispida) and, to a lesser extent, bearded seal (Erignathus barbatus) [75], primarily consuming high-calorie blubber with a specific, restricted nutritional input [76]. In contrast, onshore bears have access to a more varied but less natural diet, including bowhead whale bone piles, which can consist of whale blubber, meat, and viscera, as well the carcasses of fish, birds and caribou (Rangifer tarandus) [13, 42, 71], a more varied food source in terms of both species and tissue types.
Not only do onshore bears consume a larger range of food items, but they also likely come into contact with more terrestrial species and their associated bacteria and pathogens. Whale bone piles are utilised by a range of other nearshore/terrestrial scavengers [5, 19] providing an inter-specific focal point for many species with which polar bears do not typically interact. Beach-cast bowhead whale remains frequently lie in close proximity to settlements and towns, increasing the potential for microbiota and pathogen spillover to polar bears from humans, and domestic animals. The high gut microbiota diversity seen in onshore bears may therefore be associated with this complex network of interspecific contacts. A secondary consequence of high inter-species contact could be a higher parasite load and/or diversity in polar bears, which is associated with high gut microbiota diversity in other species [23, 29, 77].
Understanding the ways in which polar bears respond to climate-change mediated displacement from primary habitat is crucial in discerning their ability to cope with an increasingly changeable and uncertain environment [42]. Future management plans for polar bears could therefore benefit from a better understanding of the relationship between habitat availability, microbiota and health. Our results suggest that climate driven changes in land use by bears leads to changes in gut community composition, but further analyses are needed to determine whether these changes are linked to underlying causes such as diet, parasites and health. It has been suggested that researchers should incorporate health assessments into wildlife conservation practices [78, 79] and long term faecal microbiota monitoring could provide this framework.
References
Regehr EV, Laidre KL, Akçakaya HR, Amstrup SC, Atwood TC, Lunn NJ, et al. Conservation status of polar bears (Ursus maritimus) in relation to projected sea-ice declines. Biol Lett. 2016;12:20160556.
Amstrup SC, Deweaver ET, Douglas DC, Marcot BG, Durner GM, Bitz CM, et al. Greenhouse gas mitigation can reduce sea-ice loss and increase polar bear persistence. Nature. 2010;468:955–8.
Parmesan C. Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst. 2006;37:637–69.
McKinney MA, Letcher RJ, Aars J, Born EW, Branigan M, Dietz R, et al. Flame retardants and legacy contaminants in polar bears from Alaska, Canada, East Greenland and Svalbard, 2005–2008. Environ Int. 2011;37:365–74.
Atwood TC, Duncan C, Patyk KA, Nol P, Rhyan J, McCollum M, et al. Environmental and behavioral changes may influence the exposure of an Arctic apex predator to pathogens and contaminants. Sci Rep. 2017;7:13193.
Derocher AE, Aars J, Amstrup SC, Cutting A, Lunn NJ, Molnár PK, et al. Rapid ecosystem change and polar bear conservation. Conserv Lett. 2013;6:368–75.
Laidre KL, Stirling I, Lowry LF, Wiig Ø, Heide-Jørgensen MP, Ferguson SH. Quantifying the sensitivity of arctic marine mammals to climate-induced habitat change. Ecol Appl. 2008;18:S97–S125.
Regehr EV, Hunter CM, Caswell H, Amstrup SC, Stirling I. Survival and breeding of polar bears in the southern Beaufort Sea in relation to sea ice. J Anim Ecol. 2010;79:117–27.
Amstrup SC, Marcot BG, Douglas DC. A Bayesian Network Modeling Approach to Forecasting the 21st Century Worldwide Status of Polar Bears. Arctic sea ice decline: observations, projections, mechanisms, and implications. Washington DC: American Geophysical Union (AGU); 2008; pp 213–268.
Stroeve JC, Markus T, Boisvert L, Miller J, Barrett A. Changes in Arctic melt season and implications for sea ice loss. Geophys Res Lett. 2014;41:1216–25.
Stern HL, Laidre KL. Sea-ice indicators of polar bear habitat. Cryosphere. 2016;10:2027–41.
Schliebe S, Rode KD, Gleason JS, Wilder J, Proffitt K, Evans TJ, et al. Effects of sea ice extent and food availability on spatial and temporal distribution of polar bears during the fall open-water period in the Southern Beaufort Sea. Polar Biol. 2008;31:999–1010.
Herreman J, Peacock E. Polar bear use of a persistent food subsidy: Insights from non-invasive genetic sampling in Alaska. Ursus. 2013;24:148–63.
McKinney MA, Peacock E, Letcher RJ. Sea ice-associated diet change increases the levels of chlorinated and brominated contaminants in polar bears. Environ Sci Technol. 2009;43:4334–9.
McKinney MA, Stirling I, Lunn NJ, Peacock E, Letcher RJ. The role of diet on long-term concentration and pattern trends of brominated and chlorinated contaminants in western Hudson Bay polar bears, 1991–2007. Sci Total Environ. 2010;408:6210–22.
Dietz R, Riget F, Cleemann M, Aarkrog A, Johansen P, Hansen JC. Comparison of contaminants from different trophic levels and ecosystems. Sci Total Environ. 2000;245:221–31.
Hoekstra PF, O’Hara TM, Fisk AT, Borgå K, Solomon KR, Muir DCG. Trophic transfer of persistent organochlorine contaminants (OCs) within an Arctic marine food web from the southern Beaufort–Chukchi Seas. Environ Pollut. 2003;124:509–22.
Bentzen TW, Follmann EH, Amstrup SC, York GS, Wooller MJ, Muir DCG, et al. Dietary biomagnification of organochlorine contaminants in Alaskan polar bears. Can J Zool. 2008;86:177–91.
Miller S, Wilder J, Wilson RR. Polar bear–grizzly bear interactions during the autumn open-water period in Alaska. J Mammal 2015;96:1317–25.
McKinney MA, Atwood TC, Pedro S, Peacock E. Ecological change drives a decline in mercury concentrations in southern Beaufort Sea polar bears. Environ Sci Technol. 2017;51:7814–22.
Walk ST, Blum AM, Ewing SA-S, Weinstock JV, Young VB. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm Bowel Dis. 2010;16:1841–9.
Lee SC, Tang MS, Lim YAL, Choy SH, Kurtz ZD, Cox LM, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. 2014;8:e2880.
Kreisinger J, Bastien G, Hauffe HC, Marchesi J, Perkins SE. Interactions between multiple helminths and the gut microbiota in wild rodents. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140295.
Shehata AA, Schrödl W, Aldin AA, Hafez HM, Krüger M. The effect of glyphosate on potential pathogens and beneficial members of poultry microbiota in vitro. Curr Microbiol. 2013;66:350–8.
Joly C, Gay-Quéheillard J, Léké A, Chardon K, Delanaud S, Bach V, et al. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME®) and in the rat. Environ Sci Pollut Res. 2013;20:2726–34.
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–88.
Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489:231–41.
Xing M, Hou Z, Yuan J, Liu Y, Qu Y, Liu B. Taxonomic and functional metagenomic profiling of gastrointestinal tract microbiome of the farmed adult turbot (Scophthalmus maximus). FEMS Microbiol Ecol. 2013;86:432–43.
Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–23.
Pascoe EL, Hauffe HC, Marchesi JR, Perkins SE. Network analysis of gut microbiota literature: an overview of the research landscape in non-human animal studies. ISME J. 2017;11:2644–51.
Sommer F, Ståhlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, et al. The gut microbiota modulates energy metabolism in the hibernating brown bear ursus arctos. Cell Rep. 2016;14:1655–61.
Chevalier C, Stojanović O, Colin DJ, Suarez-Zamorano N, Tarallo V, Veyrat-Durebex C, et al. Gut microbiota orchestrates energy homeostasis during cold. Cell. 2015;163:1360–74.
Bestion E, Jacob S, Zinger L, Di Gesu L, Richard M, White J, et al. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat Ecol Evol. 2017;1:0161.
Harper SL, Edge VL, Schuster-Wallace CJ, Berke O, McEwen SA. Weather, water quality and infectious gastrointestinal illness in two inuit communities in nunatsiavut, canada: potential implications for climate change. Ecohealth. 2011;8:93–108.
Glad T, Bernhardsen P, Nielsen KM, Brusetti L, Andersen M, Aars J, et al. Bacterial diversity in faeces from polar bear (Ursus maritimus) in Arctic Svalbard. BMC Microbiol. 2010;10:10.
Glad T, Kristiansen VF, Nielsen KM, Brusetti L, Wright A-DG, Sundset MA. Ecological characterisation of the colonic microbiota in arctic and sub-arctic seals. Micro Ecol. 2010;60:320–30.
Xue Z, Zhang W, Wang L, Hou R, Zhang M, Fei L, et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. MBio. 2015;6:e00022–15.
Song C, Wang B, Tan J, Zhu L, Lou D, Cen X. Comparative analysis of the gut microbiota of black bears in China using high-throughput sequencing. Mol Genet Genom. 2017;292:407–14.
Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–41.
Thomas V, Clark J, Doré J. Fecal microbiota analysis: an overview of sample collection methods and sequencing strategies. Future Microbiol. 2015;10:1485–504.
Calvert W, Ramsay MA. Evaluation of age determination of polar bears by counts of cementum growth layer groups. Ursus. 1998;10:449–53.
Atwood TC, Peacock E, McKinney MA, Lillie K, Wilson R, Douglas DC, et al. Rapid environmental change drives increased land use by an Arctic marine predator. PLoS One. 2016;11:e0155932.
Cattet MR, Caulkett NA, Obbard ME, Stenhouse GB. A body-condition index for ursids. Can J Zool. 2002;80:1156–61.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4.
Albanese D, Fontana P, De Filippo C, Cavalieri D, Donati C. MICCA: a complete and accurate software for taxonomic profiling of metagenomic data. Sci Rep. 2015;5:9743.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Price MN, Dehal PS, Arkin AP. Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–50.
McMurdie PJ, Holmes S. phyloseq: An R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Hijmans RJ, Williams E and Vennes C., 2012. Geosphere: Spherical trigonometry. R package version, 753.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2018.
Ginestet C. ggplot2: Elegant Graphics for Data Analysis. J R Stat Soc Ser A (Statistics Soc) 2011;174:245–6.
Foster ZSL, Sharpton TJ, Grünwald NJ. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLOS Comput Biol. 2017;13:e1005404.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3.
Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TMS, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes. 2014;4:e121–e121.
Hildebrandt MA, Hoffmann C, Sherrill–Mix SA, Keilbaugh SA, Hamady M, Chen Y, et al. High-Fat diet determines the composition of the Murine gut microbiome independently of obesity. Gastroenterology. 2009;137:1716–24.e2.
Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T, et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol. 2006;72:1027–33.
Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108:4578–85.
Dominianni C, Sinha R, Goedert JJ, Pei Z, Yang L, Hayes RB, et al. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS One. 2015;10:e0124599.
Bennett G, Malone M, Sauther ML, Cuozzo FP, White B, Nelson KE, et al. Host age, social group, and habitat type influence the gut microbiota of wild ring-tailed lemurs (Lemur catta). Am J Prima. 2016;78:883–92.
Aivelo T, Laakkonen J, Jernvall J. Population- and individual-level dynamics of the intestinal microbiota of a small primate. Appl Environ Microbiol. 2016;82:3537–45.
Maurice CF, Knowles SC, Ladau J, Pollard KS, Fenton A, Pedersen AB, et al. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015;9:2423–34.
Amato KR, Leigh SR, Kent A, Mackie RI, Yeoman CJ, Stumpf RM, et al. The gut microbiota appears to compensate for seasonal diet variation in the wild black howler monkey (Alouatta pigra). Microb Ecol. 2015;69:434–43.
Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10.
Cadotte MW, Jonathan Davies T, Regetz J, Kembel SW, Cleland E, Oakley TH. Phylogenetic diversity metrics for ecological communities: integrating species richness, abundance and evolutionary history. Ecol Lett. 2010;13:96–105.
Berg M, Stenuit B, Ho J, Wang A, Parke C, Knight M, et al. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 2016;10:1998–2009.
Bentzen TW, Follmann EH, Amstrup SC, York GS, Wooller MJ, O’hara TM. Variation in winter diet of southern Beaufort Sea polar bears inferred from stable isotope analysis. Can J of Zool. 2007;85:596–608.
McKinney MA, Atwood TC, Iverson SJ, Peacock E. Temporal complexity of southern Beaufort Sea polar bear diets during a period of increasing land use. Ecosphere. 2017;8:e01633.
Muegge BD, Kuczynski J, Knights D, Clemente JC, Gonzalez A, Fontana L, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–4.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2013;505:559–63.
Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17:72–84.
Stirling I, Archibald WR. Aspects of predation of seals by polar bears. J Fish Res Board Can. 1977;34:1126–9.
Rode KD, Robbins CT, Nelson L, Amstrup SC. Can polar bears use terrestrial foods to offset lost ice-based hunting opportunities? Front Ecol Environ. 2015;13:138–45.
Broadhurst MJ, Ardeshir A, Kanwar B, Mirpuri J, Gundra UM, Leung JM, et al. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog. 2012;8:e1003000.
Deem SL, Karesh WB, Weisman W. Putting theory into practice: wildlife health in conservation. Conserv Biol. 2008;15:1224–33.
Patyk KA, Duncan C, Nol P, Sonne C, Laidre K, Obbard M, et al. Establishing a definition of polar bear (Ursus maritimus) health: a guide to research and management activities. Sci Total Environ. 2015;514:371–8.
Acknowledgements
We would like to thank G. Durner, A. Pagano, K. Simac, L. Peacock and T. Donnelly for capturing and sampling of polar bears, which was funded by the USGS. We are grateful to F. Albonico and the staff of the Conservation Genetics Unit at the Fondazione Edmund Mach, Italy for their training and guidance. Laboratory facilities and partial funding for the metataxonomic analyses were provided by the Fondazione E. Mach. This paper was reviewed and approved by the USGS under their Fundamental Science Practices policy (http://www.usgs.gov/fsp). Any use of trade, product or firm names is for descriptive purposes only and does not imply endorsement by the US Government. S.E.W. is supported by a NERC GW4 + Doctoral Training Partnership studentship from the Natural Environment Research Council [NE/L002434/1].
Author information
Authors and Affiliations
Contributions
Study concept and design, and acquisition, analysis and interpretation of data, drafting of figures, drafting of manuscript and writing of the manuscript by S.E.W.; study and laboratory supervision, writing of the manuscript and critical revision of the manuscript by H.C.H.; bioinformatic analysis and interpretation of data by M.J.B.; study design, field work and sample collection, data analysis, editing of manuscript, critical revision of the manuscript and obtained funding by T.C.A.; study design, editing of manuscript and critical revision of the manuscript by M.A.M.; study design and technical support by M.P.; study supervision, study concept and design, writing of manuscript and critical revision of the manuscript by S.E.P.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Watson, S.E., Hauffe, H.C., Bull, M.J. et al. Global change-driven use of onshore habitat impacts polar bear faecal microbiota. ISME J 13, 2916–2926 (2019). https://doi.org/10.1038/s41396-019-0480-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0480-2
This article is cited by
-
Gut microbiota variations in wild yellow baboons (Papio cynocephalus) are associated with sex and habitat disturbance
Scientific Reports (2024)
-
Mock community as an in situ positive control for amplicon sequencing of microbiotas from the same ecosystem
Scientific Reports (2023)
-
Gut microbiota of ring-tailed lemurs (Lemur catta) vary across natural and captive populations and correlate with environmental microbiota
Animal Microbiome (2022)
-
Distinct gut microbiomes in two polar bear subpopulations inhabiting different sea ice ecoregions
Scientific Reports (2022)
-
Host phylogeny and host ecology structure the mammalian gut microbiota at different taxonomic scales
Animal Microbiome (2021)
