





Abstract
Native American genetic ancestry has been remarkably implicated with increased risk of diverse health issues in several Mexican populations, especially in relation to the dramatic changes in environmental, dietary, and cultural settings they have recently undergone. In particular, the effects of these ecological transitions and Westernization of lifestyles have been investigated so far predominantly on Mestizo individuals. Nevertheless, indigenous groups, rather than admixed Mexicans, have plausibly retained the highest proportions of genetic components shaped by natural selection in response to the ancient milieu experienced by Mexican ancestors during their pre-Columbian evolutionary history. These formerly adaptive variants have the potential to represent the genetic determinants of some biological traits that are peculiar to Mexican people, as well as a reservoir of loci with possible biomedical relevance. To test such a hypothesis, we used genome-wide genotype data to infer the unique adaptive evolution of Native Mexican groups selected as reasonable descendants of the main pre-Columbian Mexican civilizations. A combination of haplotype-based and gene-network analyses enabled us to detect genomic signatures ascribable to polygenic adaptive traits plausibly evolved by the main genetic clusters of Mexican indigenous populations to cope with local environmental and/or cultural conditions. Some of these adaptations were found to play a role in modulating the susceptibility/resistance of these groups to certain pathological conditions, thus providing new evidence that diverse selective pressures have contributed to shape the current biological and disease-risk patterns of present-day Native and Mestizo Mexican populations.
Introduction
Mexican populations represent a paradigmatic example of human groups that have recently experienced dramatic shifts in their environmental, dietary, and cultural settings, which are expected to have had relevant social and biological consequences, especially for those communities that have maintained high proportions of Native American (NA) ancestry. In fact, NA genetic background has been implicated with increased risk of a variety of health outcomes in Mestizo (i.e., admixed) individuals, in whom the effects of recent ecological transitions and Westernization of lifestyles have been predominantly investigated (Rivera et al. 2004; Aguilar-Salinas et al. 2011; Weissglas-Volkov et al. 2013; Ko et al. 2014).
Conversely, few studies have focused so far on Native Mexican (NM) populations (Acuña-Alonzo et al. 2010; Lara-Riegos et al. 2015; Romero-Hidalgo et al. 2017), who are instead the most suitable groups to be considered for inferring the pre-Columbian evolutionary history of the ancestors of modern Mexican people (Moreno-Estrada et al. 2014; Ávila-Arcos et al. 2020). These studies revealed a complex genetic structure of the main indigenous groups that have long-inhabited present-day Mexico’s territory, which is tightly related to the well-known historical, cultural, and demographic processes they have experienced (Moreno-Estrada et al. 2014; Romero-Hidalgo et al. 2017; Ávila-Arcos et al. 2020). In particular, a northwest–southeast gradient of Mexican variation has been described, along with an overall differentiation in the proportions of ancestry components between northern, central, and southern populations, which is clearly distinguishable in both NM and geographically neighboring Mestizo groups (Rubi-Castellanos et al. 2009; Moreno-Estrada et al. 2014).
Major genetic divergence between Aridoamerican (i.e., northern) and Mesoamerican (i.e., central/southern) indigenous populations has been suggested to be ascribable also to the contrasting climate and ecological conditions characterizing these macroregions (Gorostiza et al. 2012; González-Martín et al. 2015). Nonetheless, the adaptive evolution of NM ancestors in response to local environmental and/or cultural selective pressures has been scarcely investigated so far (Ávila-Arcos et al. 2020), mainly due to the lack of large panels of whole-genome sequences (WGS), which represent the most useful data to be analyzed for drawing such inferences. Unfortunately, this has also limited the identification of the genetic determinants of important biological features of present-day NM and Mestizo people. In particular, loci underlying adaptive traits that have been shaped by natural selection in relation to the ancestral milieu of these human groups might represent an important reservoir of variants with potential biomedical relevance (Sazzini et al. 2016; Ávila-Arcos et al. 2020; Sazzini et al. 2020; Landini et al. 2021). In fact, rapid and substantial modifications that occurred in modern Mexican societies might have turned some of these biological adaptations into unfavorable (i.e., maladaptive) traits, thus contributing to differential disease susceptibility among populations with varying proportions of NA ancestry.
In the attempt to overcome the unavailability of a number of WGS useful to test such a hypothesis, we used genome-wide genotype data to infer the adaptive evolution of NM groups selected as reasonable descendants of the main pre-Columbian Mexican civilizations and previously proved to present substantial proportions of NA ancestry (Moreno-Estrada et al. 2014). For this purpose, we applied a combination of haplotype-based and gene-network analyses to the Native Mexican Diversity Panel (NMDP) data set (Moreno-Estrada et al. 2014) in order to detect genomic signatures ascribable to the occurrence of selective events under a realistic approximation of a polygenic adaptation model (Gnecchi-Ruscone et al. 2018; Sazzini et al. 2020). This enabled us to investigate a mode of action of natural selection that is supposed to have played a relevant role during human evolutionary history. Indeed, polygenic adaptation implies the evolution of adaptive traits due to natural selection acting simultaneously on multiple variants, including those at low-to-moderate frequency, which are distributed on several genes involved in the same biological function. These loci generally contribute limited phenotypic effects alone, thus exemplifying a typology of variants that are highly represented in the populations’ gene pools. Therefore, polygenic selective events are expected to have occurred more frequently than hard/soft selective sweeps (Pritchard et al. 2010; Hernandez et al. 2011). According to this approach, we identified different adaptive events peculiar to the main genetic clusters of NM populations, thus providing new evidence for diverse selective pressures having contributed to shape their current biological and disease-risk patterns.
Results
After genotypes imputation of the NMDP and pre/postimputation quality control (QC) filtering (see Materials and Methods section), we obtained a “high-density imputed data set” including 3,981,990 single nucleotide variants (SNVs) characterized for 271 individuals from 15 NM populations (supplementary table S1, Supplementary Material online). This data set was used to explore the fine-scale NM genetic structure with an unprecedented level of resolution. Predominant proportion (i.e., ≥ 90%) of NA ancestry previously reported for these groups was confirmed by ADMIXTURE analysis, with the model showing the best predictive accuracy when five population clusters (K = 5) were tested (fig. 1 and supplementary fig. S1, Supplementary Material online).
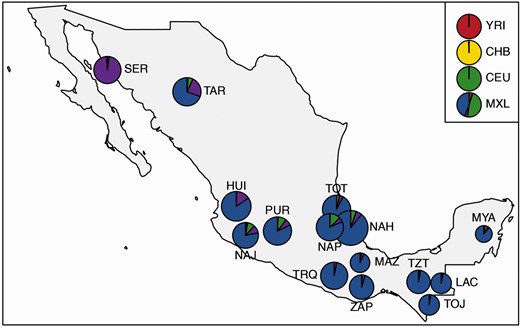
Average proportions of ancestry components inferred for the 15 Mexican indigenous groups included in the NMDP by means of ADMIXTURE unsupervised clustering analysis at K = 5. Evaluation of individual ancestry profiles was used to select subjects representative of the genomic background of their population of origin and showing predominant proportions of Native American ancestry. Individuals of African (YRI), East Asian (CHB), and European (CEU) ancestry, as well as admixed Mexican-American samples (MXL), were also included to quantify non-Native American genetic fractions observable in the indigenous genomes. SER showed a peculiar ancestry component whose high proportion was plausibly ascribable to strong genetic drift experienced due to prolonged isolation, as previously attested (Moreno-Estrada et al. 2014). Pie charts diameters are proportional to the sample sizes of the considered groups (details for each population, a full set of K tested and cross-validation errors are reported in supplementary table S1 and supplementary fig. S1, Supplementary Material online). The geographical map has been plotted using the R software (v.4.0.2).
To verify the concordance between patterns of genetic variation observed from the imputed data and those previously described for the populations examined, we first assessed NM genetic structure by performing principal component analysis (PCA) on the imputed data set, and we used Procrustes analysis to contrast it with results obtained from a PCA computed on the nonimputed NMDP. We also used the Mantel correlation test to compare identity-by-state (IBS)-based matrices of individual pairwise differences in the genome-wide proportion of shared alleles calculated on nonimputed and imputed data. As a result, the well-known northwest to a southeast cline of Mexican genetic variation was confirmed across the compared PCA plots (supplementary fig. S2, Supplementary Material online). Moreover, high and significant correlations were observed between pre and postimputation PCs (correlation in a symmetric Procrustes rotation = 0.997, P < 10−4), as well as between pre and postimputation IBS matrices (Mantel correlation = 0.84, P < 10−4).
Depicting Patterns of Haplotype Sharing among Native Mexican Populations
We used the obtained “high-density imputed data set” to explore the fine-scale genetic structure of these populations by applying the haplotype-sharing clustering approach implemented in the CHROMOPAINTER/fine-STRUCTURE pipeline. This first enabled us to detect and remove six outlier individuals that did not show close genetic affinity with those belonging to their population of origin and who may be representative of misleading mismatches between self-reported and genomic ancestry. By considering only branches of the obtained dendrogram showing posterior probability above 90%, we then identified three main clusters of NM populations: the Northern Mexican cluster (NMC), the Central Mexican cluster (CMC), and the Southern Mexican cluster (SMC) (fig. 2). This overall finding was already attested by previous studies (Gorostiza et al. 2012; Moreno-Estrada et al. 2014), but our fine-STRUCTURE analysis pinpointed a different distribution of some groups within the identified clusters. In detail, the NMC included Seri (SER) and Rarámuri (also known as Tarahumara, TAR) populations from northern Mexico. The CMC was made up of people from Central West and Central East Mexico and belonging to the Huichol (HUI), Nahua (NAH), Nahua Jalisco (NAJ), Nahua Puebla (NAP), and Purepecha (PUR) populations. The SMC grouped populations from South (i.e., Triqui, TRQ; Mazatec, MAZ; Zapotec, ZAP) and South East (i.e., Tojolabal, TOJ; Tzotzil, TZT; Lacandon, LAC; Maya, MYA) Mexico, also integrating the Totonaca (TOT) from Central East Mexico (fig. 2). An appreciable genetic differentiation was observed between NMC and CMC or SMC (Fst = 0.024, P < 10−6; Fst = 0.030, P < 10−6, respectively), although CMC and SMC turned out to be considerably less differentiated (Fst = 0.007, P < 10−6). Pairwise Fst values were computed among all populations within clusters as well (supplementary fig. S3, Supplementary Material online). A remarkable differentiation between SER and all the other NM groups was evident, with a significant Fst value obtained also for the comparison with TAR (Fst = 0.075, P < 10−6). Conversely, populations belonging to CMC and SMC showed considerably lower Fst values, with the sole exception represented by pairwise comparisons involving LAC, which were found to exceed the mean value (Fst = 0.028 ± 0.014) calculated across all possible CMC/SMC population pairs (supplementary results and supplementary fig. S3, Supplementary Material online).
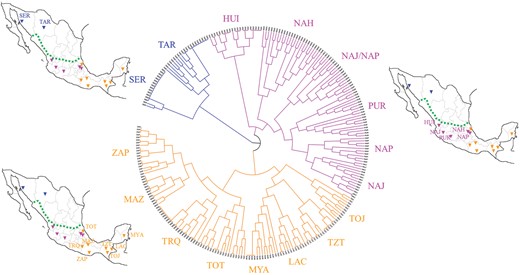
Fine-STRUCTURE hierarchical clustering dendrogram based on haplotype sharing patterns observed among the considered Native Mexican groups and their geographical locations. Population clusters were defined by considering branches of the dendrogram that split with a posterior probability above 90%. Accordingly, the Northern Mexican Cluster included SER and TAR populations, the Central Mexican Cluster was made up of groups from Central West and Central East Mexico (i.e., HUI, NAH, NAJ, NAP, and PUR), while the Southern Mexican Cluster grouped populations from Southern Mexico (i.e., TRQ, MAZ, ZAP), South East Mexico (i.e., TOJ, TZT, LAC, MYA), and Central East Mexico (i.e., TOT). The dotted line in the geographical maps indicates the demarcation between Aridoamerican (northward) and Mesoamerican (southward) regions, which represent the main geographical and cultural areas of ancient Mexico and whose population clusters showed the greatest genetic differentiation according to Fst analyses. The geographical maps have been plotted using the R software (v.4.0.2).
Differential Genomic Signatures of Positive Selection across Native Mexican Clusters
The Number of Segregating sites by Length (nSL) and the H12 statistics were calculated on the “phased nonimputed NMDP data set” for each of the identified population clusters. We then used the obtained nSL and H12 genome-wide distributions to implement a gene-network approach (see Materials and Methods section) aimed at inferring the adaptive evolution of each cluster. In particular, the most reliable selection signatures were identified as those ascribable to biological functions putatively targeted by widespread natural selection according to results from the nSL statistics that were supported also by the H12 test (see Materials and Methods section, supplementary results, Supplementary Material online). The rationale behind the independent investigation of the adaptive evolution of each cluster was that NM populations belonging to the same genetic group have presumably experienced similar environmental and/or cultural selective pressures, as conceivable according to a large body of anthropological, archeological, and historical evidence (Hard and Merrill 1992; Álvarez Palma and Casiano 2003; Báez-Jorge 2009; Rentería Valencia 2009; Ragsdale and Heather 2015; Rangel-Villalobos et al. 2016). The sole exception was represented by NMC populations (i.e., SER and TAR), who were analyzed separately due to their significant genetic differentiation and because they have long maintained different lifestyles despite having encountered similar environmental conditions in Aridoamerica (MacWilliams et al. 2008; Rentería Valencia 2009).
We also assessed whether the identified selection signatures are ascribable to adaptive events shared among populations of NA ancestry, or which are instead informative about local adaptations evolved by the studied NM groups. For this purpose, we replicated selection scans and gene-network analyses on a control cluster of South American populations that were previously proved to retain an appreciable fraction of genetic ancestry in common with Mexican people (Gnecchi-Ruscone et al. 2019, supplementary results, Supplementary Material online). We thus identified six gene networks made up of a total of 39 genes that have been significantly targeted by positive selection in this control group (supplementary table S2, Supplementary Material online), but that did not overlap with the putative adaptive loci and/or functional pathways identified for NM clusters (supplementary fig. S4, Supplementary Material online).
Seri Population
nSL-based selection scans performed on SER revealed three gene networks made up of a total of 10 genes presenting widespread signatures of positive selection (fig. 3a, supplementary table S3, Supplementary Material online). Three of these genes are implicated in the Starch and sucrose metabolism pathway (supplementary fig. S5, Supplementary Material online) and, according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, they also play a role in the Galactose metabolism and Fructose and mannose metabolism pathways by contributing to the degradation/utilization of other simple sugars. Moreover, they participate in the regulatory steps of the Glycolysis/Gluconeogenesis and Insulin signaling pathways. In particular, SI is highly expressed in the brush border of the small intestine, where the final stage of carbohydrate digestion occurs, including that of sucrose, maltose, and starch intermediates (UniProt Consortium 2019). Conversely, HK2 and PGM1 encode proteins of the hexokinase and phosphohexose mutase families that catalyze, respectively, the phosphorylation of hexoses to produce D-fructose-6-phosphate or D-glucose-6-phosphate, the first and rate-limiting step in most glucose metabolism pathways, and the phosphate transfer between positions 1 and 6 of glucose, which is critical in the breakdown and synthesis of glucose (UniProt Consortium 2019). As reported in the KEGG database, these loci are also involved in the pathogenesis of some metabolic disorders, among which type II diabetes (T2D).
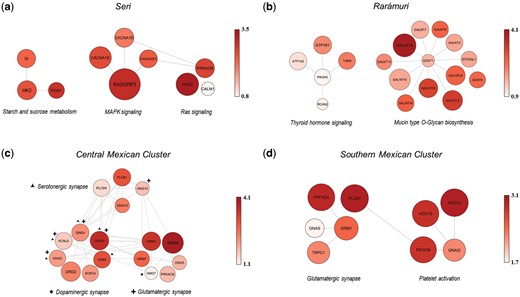
Representation of gene networks mediating polygenic adaptive events inferred for Seri, Rarámuri, and genetic clusters of Native Mexican populations identified according to fine-STRUCTURE analysis. (a) Gene networks belonging to the Starch and sucrose metabolism, Ras signaling, and MAPK signaling pathways showing significant footprints of positive selection in the SER population. (b) Gene networks belonging to the Thyroid hormone signaling and Mucin type O-Glycan biosynthesis pathways having experienced multiple selective events in the TAR population. (c) Highly interrelated gene networks belonging to the Glutamatergic, Serotonergic, and Dopaminergic synapse pathways showing widespread selection signatures in Central Mexican Cluster populations. Genes playing a role in other pathways other than the one in which they are represented are indicated with symbols: plus, Glutamatergic pathway; triangle, Serotonergic pathway; star, Dopaminergic synapse pathway. (d) Gene networks belonging to the Glutamatergic synapse and Platelet activation pathways having mediated the adaptive evolution of Southern Mexican Cluster populations. The color intensity of circles is displayed according to the reported color scale and, along with circles size, is proportional to the per-gene nSL representative scores used to perform gene-network analyses.
The other two significant gene networks pointed out by nSL-based analyses included seven genes involved in the Ras signaling and MAPK signaling pathways, which are functionally related by PRKACB (fig. 3a, supplementary fig. S6, supplementary table S3, Supplementary Material online). Signaling through the Ras/Raf/MAPK cascade responds primarily to growth factors and mitogens to ultimately regulate cell proliferation, differentiation, and apoptosis (Morrison 2012). Among the loci that showed the most relevant selection signatures in these gene networks, there was RASGRP3, which encodes a guanine nucleotide exchange factor that activates Ras proteins by switching its Ras-bound GDP to GTP, and GNG2 that encodes the gamma subunit of heterodimeric G proteins and acts as an upstream regulator of RASGRP3. Moreover, CACNA1E, CACNA1D, and CACNA2D1 encode for subunits of calcium voltage-gated channels involved in calcium-dependent processes, such as muscle contraction, hormone or neurotransmitter release, gene expression, and cell cycle (UniProt Consortium 2019) (fig. 3a, supplementary table S3, Supplementary Material online).
Results from H12-based gene network analyses indicated as possible targets of positive selection biological functions strongly related to those suggested by the nSL test. Indeed, they pointed to significant gene networks belonging to the Carbohydrate digestion and absorption and Calcium signaling pathways, with the former network including also a locus encoding for a calcium voltage-gated channel (CACNB4) similar to those previously described. By interacting with the CALM1, PRKACB, and GNG2 putative adaptive genes identified by nSL-based analyses, CACNB4 is known to participate in the Sweet taste signaling pathway as well, in which the putative adaptive genes ITPR2 and PRKG1 identified by the H12 test are also involved (supplementary table S3, Supplementary Material online).
Rarámuri Population
Two gene networks belonging to the Thyroid hormone signaling and Mucin type O-Glycan biosynthesis pathways, which included 18 genes according to nSL-based analyses and five genes according to H12-based tests, were found to have experienced multiple selective events in TAR (fig. 3b, supplementary figures S7 and S8, supplementary table S4, Supplementary Material online). The thyroid hormones (TH) T3 (triiodothyronine) and T4 (thyroxine) play an important role in the organism’s growth and development, as well as in the regulation of glucose and lipid metabolism, thermogenesis, and energy homeostasis, with T3 being considered the main active form and T4 its precursor. The identified putative adaptive genes are primarily involved in the alternative noncanonical and more rapid pathway of TH and encode for Na+/K+-ATPases (e.g., ATP1B1 and ATP1A2), TRβ isoforms (THRB), subunits of PI3K proteins (PIK3R5), and a calcineurin inhibitor (RCAN2) (fig. 3b, supplementary fig. S7, supplementary table S4, Supplementary Material online) (Moeller and Broecker-Preuss 2011). Through the noncanonical actions of TH, regulation of gene expression is achieved by an initial nontranscriptionally mediated change in cell physiology. For example, TH can induce modifications in the phosphorylation and activation patterns of several pathways, as well as in modulation of availability of second messengers and plasma membrane ion pump activity (e.g., Na+/K+-ATPase, Na+/H+ exchanger, and Ca++-ATPase) (Cordeiro et al. 2013; Senese et al. 2014). These mechanisms play a relevant role in energy metabolism by regulating glucose and triglyceride concentrations, body temperature, locomotor activity, and heart rate (Hönes et al. 2017). For instance, TRβ isoforms (THRB) are expressed mainly in cardiac and skeletal muscle, brain, and adipose tissue.
Candidate genes belonging to the Mucin type O-Glycan biosynthesis pathway instead participate in O-glycosylation of mucins, a post-translational modification mediated by glycosyltransferase enzymes and consisting in the addition of monosaccharides (O-glycans) to the hydroxyl group of serine or threonine residues. Mucins represent a family of large and heavily N-acetylgalactosamine (O-GalNAc) glycosylated proteins highly expressed by epithelial tissues that produce gel-like secretions to lubricate and protect airways, urogenital and gastrointestinal tracts against physical, chemical, or biological insults. Mucin-type O-glycans can also serve as surface receptors for adhesion molecules, mediate the interaction with pathogens including bacterial, fungal, and viral infections, and locally modulate inflammatory responses (Bergstrom and Lijun 2013; Duarte et al. 2016). Among O-glycans, O-GalNAc is especially relevant for mucin formation and genes showing selection signatures in TAR encode for several GalNAc transferases (GALNT genes) that fine-tune mucin biosynthesis (fig. 3b, supplementary fig. S8, supplementary table S4, Supplementary Material online).
Populations Belonging to the Central Mexican Cluster
In CMC populations, nSL-based analyses highlighted gene networks belonging to the Glutamatergic, Serotonergic, and Dopaminergic synapse pathways showing widespread signatures of positive selection distributed across 17 highly interrelated and overlapping loci mediating dependence on addictive substances (e.g., alcohol) (fig. 3c, supplementary figures S9–S11, supplementary table S5, Supplementary Material online). Glutamate, serotonin (5-HT), and dopamine are important neurotransmitters in the central nervous system, with glutamate representing the major excitatory one and being involved in synaptic plasticity, memory retention, and learning, although serotonin regulates behavioral and physiological functions, such as sleep and wake states, emotions, and hemostasis, among others. Dopamine, instead, controls locomotor and visceral functions, motivational, and rewarding behaviors, and has been implicated in addiction to activities and substances providing excitement and pleasure (Kanehisa and Goto 2000; Niyonambaza et al. 2019). Genes showing some of the most relevant selection signatures in CMC encode proteins that modulate the inhibitory mechanisms of these systems by suppressing the excess release of neurotransmitters. For example, the Group II (GRM2) and III (GRM7 and GRM8) metabotropic glutamate receptors (mGluRs), as well as the dopamine receptor D2 (DRD2), are Gi/o type G-protein coupled autoreceptors. They suppress excess glutamate and dopamine release via inactivation of the adenylate cyclase/cAMP/protein kinase pathway and the activation of G-protein-coupled inwardly rectifier K+ channels—GIRKs—(KCNJ3), leading to membrane hyperpolarization and suppression of neuronal excitability. Moreover, the splice variant of the G-αi2 protein (GNAI2) regulates the cell surface density of the dopamine receptor D2 by sequestrating it as an intracellular reservoir (López-Aranda et al. 2007). Among the other genes showing smoother selection signatures, some encode the beta and gamma subunits of G-proteins (GNBs and GNGs) and a member of the protein kinase cAMP family (PRKACB). PLCB1 and PLCB4 genes are instead specifically involved in the mobilization of intracellular calcium (Ca2+) stores. However, the mechanism of action of the named neurotransmitters, especially their excitatory neurotransmission, also requires intracellular Ca2+ mobilization via their coupling to G-proteins that activate phospholipases C (PLCBs) to form inositol 1,4,5-trisphosphate (IP3). IP3 then binds to the IP3R-calcium release channels receptors in the endoplasmic reticulum to increase cytosolic Ca2+ levels. When cross-checking the identified putative adaptive loci with information from the KEGG database, genes belonging to these pathways were also found to contribute to the Alcoholism, Morphine addiction, Retrograde endocannabinoid signaling, and Cocaine addiction pathways. Moreover, they were found to play a role also in immunological and inflammatory responses to viruses and trypanosomes by participating in the Human cytomegalovirus infection, Chagas disease, and Chemokine signaling pathways. Finally, also loci constituting the significant gene network pointed out by H12-based analyses belong to the Chemokine signaling pathway, with two of them (ARRB2 and GNAI2) being known to participate in the Dopaminergic synapse pathway as well (supplementary table S5, Supplementary Material online), thus confirming this biological function as a plausible target of positive selection in CMC populations.
Populations Belonging to the Southern Mexican Cluster
SMC populations showed signatures of positive selection similar to those observed for CMC groups, for instance at genes implicated in immune and inflammatory responses to viruses and trypanosomes (e.g., PLCB1 and GNAI2) (fig. 3d, supplementary table S6, Supplementary Material online). In fact, nine putative adaptive loci participate in the Glutamatergic synapse and Platelet activation pathways according to nSL-based results (supplementary figures S12 and S13, Supplementary Material online) and when we mapped them in the KEGG functional cascades, they turned out to be involved also in the Chemokine signaling, Inflammatory mediator regulation of TRP channels, Human cytomegalovirus infection, and Chagas disease pathways. This confirmed their primary role in immunity and inflammation. Indeed, although platelets have been long known to act as regulators of hemostasis and thrombosis, their additional role in innate immune reactions against bacteria, viruses, and even tumors has been recently demonstrated. They have been shown to express nine Toll-like receptors differently by gender during pathogen detection, as well as to have the ability to sample the blood environment to present viruses or bacteria to immune cells (Koupenova et al. 2015; Holinstat 2017). Functioning of the Platelet activation pathway, including granule secretion and spreading, depends on the increase of intracellular Ca2+ concentration, and the PLCB1 locus, which was found to be a putative adaptive gene also in CMC, is particularly implicated in the mobilization of intracellular Ca2+ stores and in the transduction of intracellular Ca2+ mediated signals, as is the case also for TRPC1, PPP3CA, ADCY3, and ADCY8. H12-based results confirmed genes belonging to the Glutamatergic synapse pathway as putative targets of positive selection, along with a gene network of the Chemokine signaling cascade that was made up of loci also involved in the Platelet activation pathway (e.g., GNAI2, RHOA, and ROCK1) (supplementary table S6, Supplementary Material online).
Discussion
To address the lack of studies aimed at inferring the adaptive evolution of NM groups, we focused on populations selected as reasonable descendants of the main pre-Columbian civilizations that inhabited the geographical regions nowadays attributed to the Mexican territory. For this purpose, we used the NMDP to collect data for groups showing predominant proportions (i.e., ≥ 90%) of NA ancestry and representative of the main gene pools distinguishable within the overall Mexican population (fig. 1) (Moreno-Estrada et al. 2014; Ávila-Arcos et al. 2020). However, previous studies conducted on these indigenous groups relied on DNA microarray data and/or exome variation, which prevented the exploration of a reliable proxy of the full spectrum of Mexican alleles, thus limiting the potential in investigating their fine-scale population structure. In the attempt to overcome this issue, we imputed genome-wide genotypes from the NMDP to considerably increase the density of the available genomic data set in order to use it for refining the genetic relationships between the considered NM populations.
Fine-Scale Genetic Structure of Native Mexican Populations
The fine-STRUCTURE analysis enabled us to identify three broad clusters of genetically homogenous NM populations that are expected to have shared a remarkable fraction of their evolutionary histories (fig. 2). The observed patterns of fine-scale population structure were found to be consistent with the putative ancestral geographic location of the bulk of populations that composed these genetic clusters (fig. 1, supplementary fig. S2, Supplementary Material online). TOT from Central East Mexico represented the sole exception, being settled near the Gulf Coast, but clustering within SMC, as suggested also by their partial overlap with some Southern Mexican populations in the PCA plot (supplementary fig. S2, Supplementary Material online). This peculiar outcome was likely ascribable to the presence of a geographic barrier (i.e., the Sierra Madre Oriental) that segregated TOT from the other CMC groups. Moreover, TOT has historically maintained hostile relationships with neighboring populations such as the Aztecs, who represented the ancestors of present-day Nahua people, instead of establishing intense social and commercial exchanges with southern Mexican populations (Báez-Jorge 2009). Such a genetic affinity between TOT and SMC groups is consistent also with osteological, archeological, and ethnohistorical evidence (Ragsdale and Heather 2015) and is in agreement with previous findings pointing to gene flow between TOT and MYA from South East Mexico (Moreno-Estrada et al. 2014).
Overall, a greater genetic differentiation was observed between NMC and CMC or SMC (Fst ≥ 0.024, P < 10−6) than between CMC and SMC (Fst = 0.007, P < 10−6). This suggests the main divergence between populations that have inhabited Aridoamerica and Mesoamerica, the two major geographical and cultural areas of ancient Mexico, which have been long characterized by contrasting climates and biodiversity landscapes. Such a fragmented demographic history of Mexican groups was proposed also by previous studies, which pointed to a NM genetic structure that is more related to the territories’ orography rather than to cultural/linguistic differences (Gorostiza et al. 2012; González-Martín and Gorostiza 2013; Moreno-Estrada et al. 2014; Rangel-Villalobos et al. 2016). Our results are also consistent with some of those obtained from exome data of NM populations, including TAR, HUI, TRQ, and MYA, which inferred that northern and southern ancestral groups split ∼7.2 thousand years ago (KYA) and further diverged locally around 6.5 and 5.7 KYA, respectively (Ávila-Arcos et al. 2020). More recent differentiation between HUI/PUR (3.4 KYA), NAH/PUR (2.2 KYA), and HUI/NAH (357 YA) seem to support our assumption that these CMC populations largely shared a common evolutionary history since they diverged from NMC and SMC groups (González-Martín and Gorostiza 2013). This pattern is also consistent with the frequency distribution of the four “Pan-American” mtDNA haplogroups (i.e., A2, B2, C1, D1) in Aridoamerican (e.g., Pima) and Mesoamerican indigenous groups (among which HUI, NAH, Otomí, MYA). Geographic discontinuities in their frequencies indeed revealed two major genetic boundaries deeply delimiting Aridoamericans from Mesoamerican groups, as well as a southern boundary differentiating MYA from central Mesoamerican populations (Gorostiza et al. 2012).
When considering NMC populations, a noteworthy differentiation was observed also between SER and TAR (Fst = 0.075, P < 10−6) in accordance with the lack of evidence for their historical relationships. SER has indeed shown remarkable linguistic and genetic differentiation with respect to other NM populations, including TAR, and an independent ancestry component (fig. 1, supplementary fig. S1, Supplementary Material online) suggesting a high degree of genetic drift due to prolonged isolation (Moreno-Estrada et al. 2014). Regions inhabited by SER and TAR are separated by a geographic barrier represented by the Sierra Madre Occidental and, among Aridoamericans, SER maintained a semi-nomadic hunter-gatherer lifestyle until the mid-20th century, while there is evidence for TAR ancestors having become mobile farmers since ∼3.6 KYA (MacWilliams et al. 2008; Rentería Valencia 2009). Therefore, these populations might have clustered together into NMC mainly because they were substantially differentiated from both CMC and SMC ones, rather than because they present a considerable genetic affinity.
Conversely, despite presenting the highest levels of differentiation according to pairwise Fst analyses conducted on CMC/SMC populations (supplementary fig. S3, Supplementary Material online), LAC was considered to belong to the SMC cluster. Their Fst patterns indeed plausibly reflect the effects of low population size and remarkably recent isolation (supplementary results, Supplementary Material online), as suggested also by previous studies (Moreno-Estrada et al. 2014). Therefore, we can reasonably suppose that LAC has shared most of its evolutionary history in terms of ecological and/or cultural settings with the other SMC groups.
Based on this body of evidence, we assumed that SER, TAR, CMC, and SMC populations might have adaptively evolved independently in response to differential environmental- and/or cultural-related selective pressures. Accordingly, we used the “phased nonimputed NMDP data set” to search for genomic footprints left by the action of natural selection in the identified genetically homogenous NM groups, by testing for the occurrence of selective events under a realistic approximation of a polygenic adaptation model. We thus identified weak, in terms of absolute scores per variant (supplementary table S7 and S8, Supplementary Material online), but widespread selection signatures at several functionally interconnected loci, which synergistically contributed a remarkable impact on varying biological functions across the populations under consideration.
Seri Possible Metabolic Adaptation to a Diet Rich in Simple Carbohydrates
Metabolism of simple carbohydrates (i.e., mono and disaccharides) and regulation of glucose homeostasis via the Glycolysis/Gluconeogenesis, Insulin signaling, Carbohydrate digestion, and absorption, and Sweet taste signaling pathways emerged as the biological functions that most plausibly evolved adaptively in SER (fig. 3a, supplementary fig. S5, supplementary table S3, Supplementary Material online). Selective events at genes playing a role in these metabolic processes seem to be consistent with the peculiar diet and lifestyle of this NM group, which has long relied on the consumption of fruits of succulent plants that are rich in simple sugars.
According to archeological evidence, this semi-nomadic population originally known as Comcáac has inhabited the dry desert, coastal margins, and neighboring islands of the current state of Sonora in Northwestern Mexico since at least 2 KYA. Moreover, they have maintained their hunting-gathering and fishing-based lifestyle until the 1950s (Rentería Valencia 2009). They used to organize themselves into mobile groups or “bands,” moving around in camps on the shore and islands during the dry-cold season while occupying the desert areas during the wet-hot period. During the cold season, their diet was based on the tender stem of two species of cholla cacti (i.e., Cylindropuntia bigelovii and Cylindropuntiafulgida), which provided up to 80% of the total carbohydrates consumed, as well as on hunting products and sea resources (Álvarez Palma and Casiano 2003; Hernández Santana 2017). During the hot season, they consumed mainly fruits and seeds of various species of columnar cacti, among which the saguaro (Carnegiea gigantea), sweet pitaya (Stenocereus thurberi), cardon (Pachycereus pringlei), sour pitahaya (Stenocereus gummosus), and cina cactus (Stenocereus alamosensis), from which they also prepared sweet, fermented drinks. The sweet pods from mesquite tree and the prickly pears from the cholla and nopal (Opuntia ficus-indica) were also consumed during this season (Álvarez Palma and Casiano 2003; Hernández Santana 2017). Therefore, we can speculate that selective events at multiple loci contributing mainly to sucrose, fructose, and mannose metabolism, as well as to signal transduction initiated by sweet food intake, might have optimized the utilization of their high amounts of ingested sugars as energy sources or reserves, also reducing the potential metabolic risk associated with glucose overload (Laffitte et al. 2014; Jang et al. 2018; Rabbani and Thornalley 2019). In line with such a hypothesis, among the candidate adaptive genes identified, HK2 and PGM1 encode proteins of the hexokinase and phosphohexose mutase families, respectively, involved in the rate-limiting step control of many glucose metabolism pathways, as well as in glucose breakdown and synthesis. In particular, the HK2 product was proved to have a high affinity for glucose and to be highly expressed in the skeletal muscle and adipose tissue (Tan and Miyamoto 2015). Furthermore, the SI enzyme is involved in the final digestion step of sucrose and starch intermediates such as isomaltose (UniProt Consortium 2019).
Other putative targets of selection in SER were found to belong to the Ras and MAPK signaling pathways that regulate baseline biological functions, such as cell proliferation and differentiation (fig. 3a, supplementary fig. S6, supplementary table S3, Supplementary Material online). However, it remains unclear which specific selective pressures might have triggered such adaptive events, although it is noteworthy that some of these genes (e.g., CACNA1E and CACNA1D) are expressed also in pancreatic beta cells and play a role in both the Sweet taste signaling pathway and in the functional cascade leading to insulin secretion (Vajna et al. 1998). Interestingly, these loci, as well as HK2, have been also proved to be involved in T2D pathogenesis (Muller et al. 2007; Reinbothe et al. 2013).
Semi-Nomadic Lifestyle and Exposure to Maize Mycotoxins Might Have Acted as Selective Pressures on Rarámuri
Noncanonical actions of TH that are linked to the regulation of energy metabolism (Hönes et al. 2017), as well as variation at GALNT genes controlling O-glycosylation processes during mucins biosynthesis (Bergstrom and Lijun 2013), were inferred to have been shaped by positive selection in TAR (fig. 3b, supplementary figures S7 and S8, supplementary table S4, Supplementary Material online). Selective pressures plausibly related to the peculiar mobile agricultural lifestyle adopted by the ancestors of this NM group since ∼3.6 KYA, when they first settled the mountains and canyons of the southern Chihuahua State (Northwestern Mexico) (MacWilliams et al. 2008), might have contributed to the evolution of these adaptations. Throughout this geographic area of contrasting relief, temperatures (which drop below −20°C in winter, while rising to 40°C in summer), rainfall, and vegetation, TAR are known for their traditional residential mobility and reliance on food stored year-round. Over the year, they move from primary residences on the valleys to their growing-season residences, which are associated with scattered fields for maize cultivation and depots, and to their winter locations in rock shelters located in the walls of small canyons. These seasonal shifts of residence require them to routinely walk extended distances with heavy loads from the maize depots to maintain or replenish their supplies (Hard and Merrill 1992). According to archaeological evidence, by 1.5–1 KYA TAR had already devised unique methods of maize storage, such as underground, cave niches, rectangular and cylindrical structures of stone and cement, or vases of braided grass and clay, thus confirming their strong need to store food for a long time as the main subsistence strategy (Hernández Xolocotzi 1985).
It is therefore conceivable that the detected selective events might have played a role in enhancing two major biological functions that appear to be essential for TAR. On the one hand, improvement of the regulation of energy metabolism and thermoregulation might have been useful to tolerate long-distance walks with heavy loads in such up-and-down relief and extreme climatic conditions. On the other hand, gastrointestinal protection through effective mucins formation could represent a possible adaptation to the ingestion of mycotoxins present in the stored food they relied on. In fact, through the rapid noncanonical signaling mediated by THRB, ATPases, and PIK3R5 putative adaptive genes (supplementary fig. S7, supplementary table S4, Supplementary Material online), TH has been shown to increase energy metabolism, body temperature, oxygen consumption, and locomotor activity, as well as to reduce serum triglycerides concentrations by using them as a substrate for energy production (Hönes et al. 2017; Iwen, Oelkrug, and Brabant 2018). In particular, TRβGS mice lacking canonical TH receptors signaling, but preserving their noncanonical TH actions, have shown the higher volume of oxygen uptake (VO2) and distance traveled compared to control mice (Hönes et al. 2017). Such a hypothesis of enhanced energy metabolism of TAR is in line with that proposed by two previous studies reporting an enrichment of genes involved in musculoskeletal functions harboring novel missense/promoter region variants (Romero-Hidalgo et al. 2017) and a candidate adaptive gene (BCL2L13) highly expressed in skeletal muscle (Ávila-Arcos et al. 2020). Both findings suggested adaptive musculoskeletal traits underlying the well-known physical endurance of TAR.
Finally, studies evaluating the effects of exposure to mycotoxins, such as those frequently found in maize (e.g., deoxynivalenol, zearalenone, fumonisin B1, and aflatoxin B1) in relation to its storage conditions, have demonstrated a marked disruption of intestinal epithelial cells and mucus barrier integrity/function (Robert et al. 2017). The assessment of the effects exerted by deoxynivalenol and fumonisin B1 on mucus has revealed that they modulate both mucin production in the duodenum and its monosaccharide composition (namely of galactose and N-acetyl-neuraminic acid, GalNAc) by regulating the expression of genes involved in mucin biosynthesis (Wan et al. 2014; Pinton et al. 2015). Since TAR derives approximately 75% of their diet from maize and has relied mostly on stored maize (Hard and Merrill 1992), it is likely that they have been long exposed to mycotoxins ingestion, which might represent one of the selective pressures able to modulate enhanced mucin biosynthesis aimed at ensuring gastrointestinal homeostasis.
Potential Adaptation of Central Native Mexican Populations to Addictive Substances
CMC populations were supposed to have evolved adaptations mostly mediated by functional pathways involved in the dependence on addictive substances, among which alcohol, morphine, and cocaine (fig. 3c, supplementary figs. S9–S11, supplementary table S5, Supplementary Material online). Indeed, the most plausible candidate adaptive genes identified in this group encode for mGluRs (i.e., GRM2, GRM7, and GRM8) and dopamine (DRD2) autoreceptors, as well as for a protein (i.e., GNAI2) that regulates DRD2 cell surface density. These loci spearhead the inhibitory mechanisms of the related neurotransmission systems and have been associated with addictive phenotypes (Abrahao et al. 2017; Roberto and Varodayan 2017). These findings seem to be consistent with the traditional consumption of alcohol and psychoactive plants by the ancestors of these NM populations (Roman et al. 2013). Accordingly, we can hypothesize that adaptive changes at the Glutamatergic, Serotonergic, and Dopaminergic synapse pathways might have contributed to enhance biological tolerance towards such stimulant compounds and their detrimental effects, which in turn could have led to increased risk to develop a phenotype of dependence, especially after the recent cultural transition and Westernization of lifestyles occurred in modern Mexican societies. Ethno-historical evidence suggests that routine consumption of stimulant substances by the ancient Wixárika (HUI) and Aztec (Nahua) populations followed the processes of plants domestications in Mesoamerica ∼5 KYA. During this period, they also incorporated into their culture the use of fermented alcoholic beverages made from maize (tejuino) and the agave plant (“pulque”), respectively, which were endemic to this region. Alcoholic beverages were regularly consumed for religious, nutritional, and medicinal purposes, to the extent that excessive drinking leading to drunkenness became strictly prohibited and penalized by death (Roman et al. 2013). Interestingly, most of the identified putative adaptive genes are linked to the ethanol-induced changes in interneuron function and synaptic transmission associated with chronic rather than acute alcohol exposure and play a role in the development of ethanol-seeking behavior and alcohol use disorders. For example, neuroadaptations of glutamatergic transmission due to chronic ethanol exposure (CEE) and withdrawal include the enhanced function of ionotropic (iGluRs) and mGluRs glutamate receptors, resulting in increased release but decreased reuptake of glutamate in several brain regions and an extracellular hyperglutamatergic state (Roberto and Varodayan 2017). Downregulation of mGluR2 encoded by GRM2 in nucleus accumbens (NAc) neurons contributed to the hyperglutamatergic state and has been implicated in alcohol dependence (Meinhardt et al. 2013). CEE also results in diminished serotonergic and dopaminergic transmission through variable effects on 5-HT receptors, hypoactivity of dopamine neurons, and decreased NAc/striatum dopamine release and dopamine receptor D2 availability (Abrahao et al. 2017). In addition, in vivo studies have revealed that dopamine receptor D2/GIRK-mediated autoinhibition is uniquely regulated by a Ca2+-dependent desensitization mechanism involving IP3-induced Ca2+ release and stimulated by mGluRs activation (Perra et al. 2011). Direct molecular targets of ethanol include specific ion channels, such as the GIRKs, encoded by the identified putative adaptive KCNJ3 gene, as well as nonion-channel targets, such as intracellular signaling molecules encoded for example by the PRKACB locus (fig. 3c, supplementary table S5, Supplementary Material online) (Abrahao et al. 2017). Moreover, the HSD17B11 (a short-chain alcohol dehydrogenase that metabolizes secondary alcohols and ketones) and KCNC2 genes have been recently identified as targets of natural selection in HUI (Ávila-Arcos et al. 2020). This may support the hypothesis that traditional consumption of stimulant substances, particularly alcohol, have had the potential to play a role in the evolution of these adaptive traits. This seems to be in line also with findings showing that Nahua and HUI exhibit one of the highest frequencies documented to date (67% and 65%, respectively) of the DRD2/ANKK1 TaqIA A1 allele (rs1800497), which is associated with diminished dopamine receptor D2 expression and alcohol dependence (Panduro et al. 2017). Interestingly, in the admixed Mexican population from the same geographic region, the A1 allele was associated with heavy alcohol drinking, overconsumption of palatable foods known to have addictive properties (e.g., meat, fried dishes, and sugars), as well as increased risk of impaired glucose, triglycerides, and VLDL levels (Rivera-Iñiguez et al. 2019). Moreover, the C allele of the DRD2 C957T substitution (rs6277) was associated with increased consumption of sugar and serum triglycerides (Ramos-Lopez et al. 2018). Although the described genetic signatures overall suggest a possible ethanol-driven selective pressure, some of the identified putative adaptive genes (e.g., PLCB1 and PLCB4), along with some loci that were found to have evolved adaptively in SMC (e.g., ADCY3, TRPC1, and PPP3CA), also regulate intracellular Ca2+ mobilization and are implicated in immunological and inflammatory responses to cytomegalovirus and trypanosomes. In fact, parasites such as the American Trypanosoma cruzi and Leishmania mexicana are quite common in the central-southern regions of Mexico, so we cannot rule out the hypothesis that pathogen-related selective pressures have also contributed to trigger the inferred adaptive events.
Adaptations of Southern Native Mexican Populations Plausibly Triggered by Endemic Pathogens
SMC populations were found to share an appreciable fraction of their adaptive evolution with CMC groups, specifically due to the putative adaptive genes described above and controlling intracellular Ca2+ mobilization, which exerts crucial functions in both the Glutamatergic synapse and Platelet activation pathways (fig. 3d, supplementary figs. S12 and S13, supplementary table S6, Supplementary Material online). Nonetheless, all these loci are implicated also in immune and inflammatory responses, especially to cytomegalovirus and T. cruzi. Therefore, we can hypothesize that these (or similar) pathogens have plausibly represented some of the main selective pressures having acted on the ancestors of these NM populations, who have inhabited for millennia the coastal areas of southern Mexico, or the central-eastern coast in the case of TOT. These regions are characterized by a tropical savanna climate and represent endemic areas of parasites such as T. cruzi and L. mexicana, which are responsible for Chagas disease and localized cutaneous leishmaniasis (LCL) (Rojo-Medina et al. 2018; Loría-Cervera et al. 2019).
Interestingly, it has been demonstrated that platelets, besides playing a fundamental role in coagulation and vascular integrity, are also capable of controlling and modulating innate immune responses against pathogens, thus contributing to their clearance. This is mainly due to their abundance of membrane receptors, such as Toll-like receptors, which are able to detect pathogen- and damage-associated molecular patterns (Ribeiro et al. 2019). In line with these findings and with the hypothesis of SMC adaptations to pathogens, a recent study identified three innate immune pathways, among which that of Toll-like receptor signaling (to which the putative adaptive gene PIK3CB identified here belongs) showing evidence of NA ancestry enrichment polygenic adaptation in an admixed population of Mayan ancestry (Norris et al. 2020). Platelet granule secretion and spread depend on increased intracellular Ca2+ concentration and the ADCY3, TRPC1, PPP3CA, and PLCB1 genes that exhibited some of the most relevant selection signatures in SMC populations (fig. 3d, supplementary figs. S12 and S13, Supplementary Material online) are involved in the mobilization of intracellular Ca2+ stores. Furthermore, calcium signaling is vital for host cell invasion and the life cycle of both T. cruzi and L. mexicana parasites. Cell invasion by T. cruzi is Ca2+ dependent and its infective trypomastigote stages are capable of triggering intracellular free Ca2+ transients in its target cells (Docampo and Huang 2015; Hashimoto et al. 2016). Infection by these parasites causes a peculiar clinical picture in southern Mexican populations. For example, in a 7-year longitudinal study conducted on subjects of southern Mexican ancestry chronically affected by Chagas disease, it was unexpectedly found that there was no progression of the main Chagas-related cardiomyopathies and no chagasic mega viscera compared to the chronic clinical picture shown by subjects from different regions of South America (Goldsmith et al. 1986). Similarly, variability of the clinical presentation of LCL has been observed, with populations from endemic areas often manifesting asymptomatic infection, suggesting a host-specific immune response capable of controlling parasite replication (Andrade-Narvaez et al. 2016). Taken together, this evidence and the findings obtained by gene network analyses for SMC populations suggest that they might have evolved adaptations in response to pathogens that are endemic to the regions long inhabited by their ancestors, possibly resulting in the enhancement of their innate immune-mediated mechanisms of clearance of infection.
Conclusions
Although imputed data cannot provide the same amount and quality of information ensured by WGS, imputation of high-density genomic data for 271 individuals from 15 Mexican indigenous groups showing high proportions of NA ancestry enabled us to explore patterns of fine-scale population structure among Mexican people. This approach was essential to guide the subsequent reconstruction of the unique adaptive histories of their ancestors, which was instead carried out by using nonimputed genome-wide genotype data. Most of the loci constituting the gene networks putatively targeted by widespread natural selection were found to be distributed on different chromosomes or to be so distant within chromosomes that probably low linkage disequilibrium (LD) exists between them. Even in the very few cases in which a reduced distance between adaptive loci was attested, moderate LD values (i.e., ≤ 0.7) were observed. This suggests that the obtained results were not biased by issues related to the occurrence of dependent observations, instead of representing reliable signatures of polygenic adaptive events.
Overall, we provide new and robust evidence on the different selective pressures that have plausibly targeted the main genetic clusters identifiable within the overall Mexican population. Moreover, most of the inferred adaptive events pointed to natural selection having acted especially on the upstream part of the targeted functional pathways, as observed when assessing the location of the putative adaptive genes within the overall pathways they belong to (supplementary figs. S5–S13, Supplementary Material online). This suggests that these biological adaptions were achieved via modulation of a few rate-limiting steps that represent key control points of the related pathways, thus enabling natural selection to effectively impact a pathway's activity without completely reshaping it. This is a crucial aspect of the described adaptive processes that have been instrumental in ensuring the evolution of relatively rapid genetic adaptations, which would otherwise have been unlikely in a species characterized by low effective population sizes and reduced genetic variability as humans. Some of the identified biological adaptations were also found to have the potential to play a role in modulating susceptibility/resistance to certain pathological conditions. For instance, T2D in SER, impairment of gastrointestinal homeostasis after ingestion of mycotoxins in TAR, dependence on addictive substances/foods in CMC groups, and severity of Chagas-related comorbidities and infection by L. mexicana in SMC people. Accordingly, these findings represent valuable insights into the origins and the determinants of some biological traits that are peculiar to both NM and Mestizo people. Therefore, the obtained results might lay the foundation for future studies hopefully based on large WGS panels and aimed at devising regionalized/personalized preventive strategies properly tailored to these human groups, which is considered the best course of action to cope with the health issues related to the ecological and cultural transitions recently experienced by Mexican populations (Ojeda-Granados et al. 2017; 2020).
Materials and Methods
Analyzed Data Set
Data analyzed in the present study were retrieved from those of the NMDP, which were previously generated as described in Moreno-Estrada et al. (2014). This data set included 350 samples from 15 NM populations characterized for 903,800 genetic variants annotated according to the human reference genome build 36 (hg18). Two populations were from Northern Mexico (i.e., SER, TAR), six were from Central Mexico (i.e., HUI, NAH, NAJ, NAP, PUR, TOT), and seven were representative of people from Southern Mexico (i.e., MYA, LAC, TOJ, TZT, ZAP, MAZ, TRQ).
Genotypes Imputation
To generate a high-density genomic data set to be used for implementing fine-scale population structure analyses, the NMDP was submitted to genotype imputation. We first applied preliminary QC procedures using PLINK v2.0 (Chang et al. 2015) to retain only autosomal loci, as well as variants and samples showing genotyping success rate >95%. This led to a filtered data set made up of 347 samples and 779,283 loci, whose genomic coordinates were reannotated according to the human reference genome build 37 (hg19) using the liftOver tool and the hg18ToHg19.over.chain.gz assembly conversion files (Hinrichs et al. 2006). We then phased this data set with the Eagle v2.3 software (Loh et al. 2016) implemented in the Michigan Imputation Server (MIS) v1.0.4 (Das et al. 2016) and we used the MIS Minimac3 algorithm to impute genotypes using the 1000 Genomes Project Phase 3 as reference panel (1000 Genomes Project Consortium et al. 2015). The imputed data set was subsequently filtered to retain only high-quality genotypes by using the bcftools v1.8 package (Li et al. 2009) to remove variants showing a squared Pearson correlation coefficient (r2) ≤ 0.95 between imputed genotype probability and true genotype call, as suggested in Palmer and Pe’er (2016). This generated a data set including 347 samples and 5,481,422 SNVs.
Data Curation
The imputed data set was subjected to additional QC filters using PLINK v2.0 to retain SNVs/samples showing genotyping success rate > 95% and variants with no deviations from the Hardy-Weinberg Equilibrium (P > 1.5 × 10−9 after Bonferroni correction). In addition, we filtered out SNVs with ambiguous A/T or G/C alleles and multiallelic variants. The degree of recent shared ancestry (i.e., identity-by-descent [IBD]) for each pair of subjects was also obtained to remove closely related individuals to the second degree. IBS was calculated and used to estimate an IBD kinship coefficient (PI_HAT). According to the history of founder events, long-term isolation, low effective population size, and high inbreeding levels of NA populations, which are thus expected to present remarkable mean IBD values, a higher than usual IBD threshold of PI_HAT ≥ 0.270 was considered prompting the exclusion of 76 samples. This led to the generation of a final “high-density imputed data set” including 3,981,990 SNVs and 271 individuals.
A LD-pruned version of this data set was also created by removing one SNV for each pair showing r2 > 0.1 within sliding windows of 50 SNVs and advancing by five SNVs at the time. Genotype-based population structure analyses were thus used to check for consistency between the imputed and nonimputed NMDP data. For this purpose, we filtered the LD-pruned data set for variants with MAF < 0.05. Then, a PCA was computed using the smartpca method implemented in the EIGENSOFT package v6.0.1 (Patterson et al. 2006), while the ADMIXTURE algorithm (Alexander et al. 2009) was applied to test K = 2 to K = 12 population clusters by running 25 replicates with different random seeds for each K and by retaining only those presenting the highest log-likelihood values. Cross-validation errors were also calculated for each K to identify the value that best fit the data. Moreover, PCA and IBS-based matrices of individual pairwise differences in the genome-wide proportion of shared alleles were computed on both the nonimputed and imputed LD-pruned data sets, being then compared by means respectively of Procrustes analysis (Wang et al. 2010) and Mantel correlation test (Mantel 1967) using functions implemented in the R vegan package. Statistical significance of the obtained Mantel correlation was empirically tested over 10,000 permutations.
To perform fine-scale population structure analyses the final “high-density imputed data set” was then phased with SHAPEIT2 v2.r790 (Delaneau et al. 2013) by applying default parameters and by using HapMap phase 3 recombination maps and the 1000 Genomes Project data set as a reference panel (1000 Genomes Project Consortium et al. 2015). Conversely, to perform haplotype-based selection scans that generate a score for each of the considered genetic variants, the QC filtered nonimputed NMDP data set was similarly phased with SHAPEIT2 v2.r790 (Delaneau et al. 2013) in order to calculate selection statistics on genotyped loci and not on imputed ones. In doing so, information about ancestral/derived alleles as specified in the reconstructed reference human genome sequence (1000 Genomes Project Consortium et al. 2015) was also considered, leading to the generation of a “phased nonimputed NMDP data set.”
CHROMOPAINTER/fine-STRUCTURE Clustering Analyses
To identify groups of genetically homogenous NM populations and to remove outlier individuals not representative of their population of origin, we applied the haplotype sharing clustering methods implemented in the CHROMOPAINTER/fine-STRUCTURE package (Lawson et al. 2012) to the phased “high-density imputed data set.” The ChomoPainter v2 algorithm was used to produce a coancestry matrix through the reconstruction of haplotype sharing patterns of each individual performed by considering all other subjects in the data set as potential “donors” of DNA sequence chunks, but excluding themselves (Lawson et al. 2012). To this end, ten steps of the Expectation-Maximization algorithm were applied to a subset of chromosomes {4,10,15,22} to estimate recombination/switch rate (-n) and mutation/emission probability (-M). The obtained values (-n = 757.576090186 and -M = 0.00258239725762) were averaged across the subset of chromosomes, by weighting them by the number of SNVs, and across individuals. Mean recombination/switch rate and mutation/emission probability values were then used to run again the ChomoPainter v2 algorithm on all autosomes by considering k = 50, which is a value preferable when examining closely related populations, to specify the number of expected haplotype chunks used to define a genomic region. We thus obtained a coancestry matrix by considering the matrix of counts of haplotype chunks shared among subjects, which were then summed across all autosomes. The matrix was submitted to the Markov chain Monte Carlo (MCMC) fine-STRUCTURE v.fs2.1 algorithm (Lawson et al. 2012) by setting 1,000,000 burn-in iterations, followed by 1,000,000 more iterations and the sampling of inferred clustering patterns every 10,000 runs. A further fine-STRUCTURE v.fs2.1 run consisted of 100,000 additional hill-climbing iteration steps aimed at improving posterior probability and at merging the identified clusters in a stepwise fashion. The inferred haplotype sharing patterns were finally visualized as a circular dendrogram with the iTOL v5 tool (Letunic and Bork 2019), with population clades being defined by considering only those branches that were characterized by a posterior probability >90%.
Pairwise Fst values between population clusters were also computed according to Hudson’s method using the EIGENSOFT package v6.0.1 (Patterson et al. 2006). The calculation was replicated after LD-pruning this data set with PLINK v2.0 as previously described to maintain only variants in linkage equilibrium with each other and by using functions implemented in the R StAMPP package. This enabled us to perform 1,000 bootstraps to obtain confidence intervals and corresponding P-values associated with the computed Fst values.
Number of Segregating Sites by Length and H12 Statistics
Based on the assumption that the ancestors of NM people belonging to the same genetically homogeneous population cluster have plausibly shared a remarkable fraction of their evolutionary histories, having also experienced similar environmental- and/or cultural-related selective pressures, we searched for genomic signatures of natural selection independently in each of the identified NM genetic clusters.
For this purpose, we filtered out from the “phased nonimputed NMDP data set” the closely related/outlier individuals identified as described above and we used it to compute the nSL statistic and the H12 statistic. These tests were developed to detect a broad range of selective events ranging from the strong positive selection on newly arisen alleles to more slight selection on multiple alleles, including standing variation (Ferrer-Admetlla et al. 2014; Garud et al. 2015). This improved the exploration of the full spectrum of adaptive events evolved by NM groups, including those mediated by genetic variants with a small individual effect size. nSL scores for each genotyped variant were obtained through the application of algorithms implemented in the selscan v.1.1.0b program (Szpiech and Hernandez 2014) by setting a 20,000 bp threshold for gap scale, 200,000 bp as the maximum length a gap can reach, and 25 consecutive loci as the maximum extension parameter. H12 scores for each genotyped variant were instead computed by means of an ad hoc developed script by following Garud et al. (2015) recommendations and by using sliding windows of 200,000 bp and advancing by one variant at the time.
The obtained unstandardized nSL and H12 scores were finally normalized across 100 frequency bins by subtracting to each value the average score computed for the considered bin and by dividing the resulting score by the related standard deviation (Piras et al. 2012).
Identifying Gene Networks Showing Widespread Selective Events
To test the occurrence of selective events under a realistic approximation of a polygenic adaptation model, we performed gene-network analyses by applying the signet algorithm implemented in the R package (Gouyet al. 2017). For this purpose, we used the genome-wide distributions of normalized nSL and H12 scores previously obtained for each population cluster to associate values to each gene annotated on the human genome by considering variants within a range of 50 kb upstream and downstream of the chromosomal location of the genes. Then, we selected the highest nSL and H12 scores among those computed for all variants ascribable to the same gene as the representative score of that given genetic locus.
Per-gene scores were used as input for running the signet algorithm based on results from the nSL test, which was the statistic that we considered more suitable for informing gene-network analyses according to its power in detecting selection on standing variation and its robustness to variation in local recombination rates and demographic scenarios such as bottlenecks and population substructure (Ferrer-Admetlla et al. 2014). Then, the signet algorithm was applied also to H12 scores, to validate biological functions putatively targeted by widespread natural selection according to results from nSL-informed gene networks.
In detail, the signet algorithm concomitantly considered the information available for all biological pathways annotated in the National Cancer Institute/Nature Pathway Interaction Database to map the reconstructed gene networks to known functional pathways (Schaefer et al. 2009). In fact, the assumption behind this approach was that multiple genes within a pathway and contributing to a certain biological function might interact to shape a given adaptive phenotype, thus being targeted simultaneously by natural selection. Therefore, by using a simulated annealing approach the signet algorithm searches for networks of genes that directly interact with each other and that at the same time present widespread selection signatures (i.e., those whose distribution of scores within the pathway they belong to was shifted significantly towards extreme values). To this end, we set an iterative process of 20,000 iterations, and we generated null distributions of the highest scoring subnetwork (HSS) for each network of a specific size finally comparing the obtained HSS with them. Gene networks showing P-values < 0.05 after controlling false discovery rate (FDR) and belonging to biological functions supported by both nSL-based and H12-based analyses were considered as significantly targeted by selective events that occurred at multiple genes contributing to the same functional pathway and were plotted with Cytoscape v3.6.0 (Shannon et al. 2003). Moreover, we also performed a cross-check of the identified gene networks using the KEGG database (Kanehisa and Goto 2000) to identify additional pathways in which the putative adaptive genes might play an important role. Overall, this enabled us to shortlist the set of genes and functional pathways that have been more plausibly subjected to pervasive positive selection in the different NM population clusters.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Data Availability
The individual-level genotype data reanalyzed in the present study are available through a data access agreement by contacting the corresponding authors of the original source of the data as stated in Moreno-Estrada el al. 2014. All scripts specifically developed for the purposes of the present study are available at https://github.com/paoloabondio/Ojeda-Granados_et_al_2021 (last accessed September 3, 2021).
Acknowledgments
We would like to thank all donors who originally contributed biological samples for the generation of the NMDP without whom this work would have not been possible. We are also grateful to Pier Massimo Zambonelli (CESIA, University of Bologna) for his IT assistance. C.O.G. was supported by two postdoctoral grants (grant n. 755288, n. 711247) from the National Council of Science and Technology (CONACyT) of Mexico in collaboration with the Ph.D Program in Molecular Biology in Medicine of the University of Guadalajara (CONACyT PNPC 000091) and the Ph.D Program in Integrative Biology, CINVESTAV, Irapuato (CONACyT PNPC 003649). S.D.F. was supported by the FONDAZIONE CASSA DI RISPARMIO IN BOLOGNA (grant n. 2019.0552 to M.S.) and A.M.E. was supported by CONACyT (grant n. FONCICYT/50/2016) and The Newton Fund (grant n. MR/N028937/1).

{kind=link}
{kind=link}
{kind=link}