
Abstract
The present study aims to investigate the in vitro intestinal digestion fate of Pickering emulsions with complex dual particle interfaces. Pickering oil-in-water emulsions (PPM-E) stabilized by plant (pea) protein-based microgels (PPM), as well as PPM-E where the interface was additionally covered by cellulose nanocrystals (CNC), were designed at acidic pH (pH 3.0). The gastrointestinal fate of the PPM-E and free fatty acid (FFA) release, was tested via the INFOGEST static in vitro digestion model and data was fitted using theoretical models. Lipid digestion was also monitored using lipase alone bypassing the gastric phase to understand the impact of proteolysis on FFA release. Coalescence was observed in the PPM-stabilized emulsions in the gastric phase, but not in those co-stabilized by CNC. However, coalescence occurred during the intestinal digestion stage, irrespective of the CNC concentration added (1–3 wt % CNC). The presence of CNC lowered the lipolysis kinetics but raised the extent of FFA release as compared to in its absence (p < 0.05), due to lower levels of gastric coalescence, i.e., a higher interfacial area. The trends were similar when just lipase was added with no prior gastric phase, although the extent and rate of FFA release was reduced in all emulsions, highlighting the importance of prior proteolysis in lipolysis of such systems. In summary, an electrostatically self-assembled interfacial structure of two types of oppositely-charged particles (at gastric pH) might be a useful strategy to enable enhanced delivery of lipophilic compounds that require protection in the stomach but release in the intestines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oil-in-water emulsions are omnipresent in foods such as milk alternatives, cream, sauces, butter and functional beverages. Colloidal dispersions can be considered as an extremely efficient delivery system for macronutrients, micronutrients and nutraceuticals– including lipids, fat-soluble vitamins, omega-3 fatty acids, proteins and minerals [1,2,3,4,5]. However, many conventional emulsions suffer from physicochemical instability and concomitant variations in texture, appearance and bioavailability of the intrinsic components due to changes in temperature, pH extremes and high salt concentrations. Consequently, there has been a growing interest in designing emulsions with enhanced and/or more controlled stability. Paramount amongst these developments has been the expansion of the range of particle-stabilized emulsions (Pickering emulsions), due to their higher resistance to coalescence and Ostwald ripening [6,7,8,9] plus additional opportunities for using such emulsions to develop novel ways of protecting and delivering bioactive species [9,10,11].
Sarkar et al. [12] designed whey protein microgel particles-laden Pickering oil-in-water (O/W) emulsions and determined their ability to control the extent and rate of lipolysis using INFOGEST in vitro gastrointestinal digestion models. Shimoni et al. [13] and Meshulam and Lesmes [14] focused on lactoferrin nanoparticles (LFnp) as stabilizer and showed the influence of LFnp on O/W droplet stability during in vitro oral, gastric and intestinal conditions. These studies show promise of animal protein-based Pickering particles not only to design coalescence-resilient emulsions but also to control and mostly reduce the rate and degree of free fatty acid release during intestinal digestion.
With growing consumer demand for more plant-based food products, there has been an increasing attention on plant protein-based particles, such as soy glycinin [15], soy protein nanoparticles [16], kafirin nanoparticles [17], zein colloidal particles (ZCPs) [18, 19], peanut protein nanoparticles [20], and pea protein particles [21, 22] as Pickering emulsion stabilizers. Although Pickering emulsions, in general, have proved to be more resistant to lipolysis due to the high energy barrier to their displacement from the interface by bile salts, studies using plant protein-based microgel particles are relatively scarce [23]. Any emulsions stabilized by protein particles tend to destabilize under gastric conditions, due the low pH, mucins and/or pepsin action, making them unsuitable for site-dependent release of hydrophobic bioactives to the intestines. For this reason, combinations of stabilizers have been tested to try and improve the stimuli-responsiveness of the Pickering emulsions to enable the release of lipid-soluble nutrients like \(\beta\)- carotene and curcumin [13, 14, 18, 19, 24,25,26].
Meshulam and Lesmes [14] found that the combining of LFnp-stabilized emulsions with alginate reduced the extent and reaction rate of lipolysis, while free fatty acids (FFAs) release was enhanced in the case of the emulsions mixed with iota-carrageenan. Wei et al. [18] reported that the bioaccessibility of \(\beta\)-carotene ranged from 9.1 to 27.2% while the extent of FFA release ranged from 19.5 to 12.9% on adjusting the mass ratio of hydrophilic cellulose nanocrystals (CNC) to ZCP-stabilized Pickering emulsions. Apart from the above mentioned examples, it seems that there have been a limited number of studies of the digestion of Pickering O/W emulsions stabilized by combinations of different particles. Such combination of particles offer complex interfaces might provide an improved route to create O/W emulsion based encapsulation systems for bioactive compounds based on plant biopolymers, since many plant storage proteins and polysaccharides are relatively insoluble in the first place.
Consequently, the aim of this study was to investigate the stability of an O/W emulsion stabilized by plant-based pea protein microgels + CNC and understand the influence that this dual particle system has on FFA release from the emulsion when passing through in vitro intestinal digestion conditions without/ with a prior gastric digestion step. The thermally-cross-linked-sheared pea protein microgel particles (PPM), produced via a top down approach, have already proven their ability to serve as soft Pickering-like stabilizers of O/W emulsions [27]. These Pickering emulsions (PPM-E) showed high stability to varying temperature, pH and ionic strength. However, not surprisingly, destabilization occurred in the presence of pepsin at low pH, i.e., simulated gastric conditions [22]. It was demonstrated that the addition of unmodified CNC at pH 3.0 led to the formation of a stronger and more coherent interfacial layer, proven via Langmuir trough experiments [22]. The increased strength was derived from electrostatic attraction between the positively charged PPM and the negatively charged CNC at this pH. The negative charge on the CNC is a result of their production via sulfuric acid hydrolysis of cellulose and subsequent grafting of sulfate groups to the crystals [28,29,30,31].
Our hypothesis is that such particle–particle interactions at the interface should be broken during the following intestinal digestion stage, since this occurs at around pH 7.0, when both types of particle are negatively charged. Whether such break down of the electrostatic complexation between the particles at the interface leads to greater or lesser extent of lipolysis and consequently FFA release compared to in the absence of CNC remains to be revealed and this is the key novelty of this work. The size distributions and zeta-potentials of microgels and emulsions were measured via static light scattering techniques. Confocal laser scanning microscopy (CLSM) was employed to observe the evolution of microstructure during in vitro gastrointestinal digestion and a pH–stat-based titrimetric method was used to measure the kinetics of FFA release. In some cases, thermal treatment of the emulsions prior to gastrointestinal digestion was monitored to see if fusion of microgel particles at the interface had any influence, since this is of relevance to real food processing.
Material and Methods
Materials
Pea protein concentrate (Nutralys S85X, 85 wt% protein) was a gift from Roquette, Lestrem, France. Commercially available cellulose nanocrystals (CNC) were purchased from Celluforce™ (Quebec, Canada); this was sulfated during the processing by the manufacturer and was used without any modification. Whey protein isolate (WPI, 90 wt% protein) was gifted by Fonterra Co-operative Group Limited, Auckland, New Zealand. For the oil phase, sunflower oil was purchased from a local supermarket (Tesco, UK) and used without any additional purification. Porcine pepsin (product number: P7000, activity 650 U/mg [22]), porcine pancreatin (product number: P7545, 8 × USP, lipase activity of 82.62 U mg−1 [32, 33]), lipase from porcine pancreas Type II (activity 68 U/mg containing amylase and protease activity) and porcine bile extract (product number: B8631; total bile salt content was ~50 wt% containing glycodeoxycholic acid (10–15 wt%), taurodeoxycholic acid (3–9 wt%), deoxcycholic acid (0.5–7 wt%) and phospholipids (5 wt%) were purchased from Sigma-Aldrich Company Ltd, Dorset, UK. Analytical grade chemicals were purchased from Sigma-Aldrich (Dorset, UK), unless otherwise specified. Milli-Q water (purified by a Milli-Q apparatus, Millipore Crop., Bedford, MA, USA) was used to prepare solvents such as buffer, gastric and intestinal fluids.
Fabrication of Pea Protein Microgel Particles (PPM)
The fabrication of pea protein microgels (PPM) was carried out using a top-down method as reported previously by Zhang and coworkers [22, 27]. Briefly, heat-set hydrogels were prepared by dispersing pea protein concentrate (12.54 wt% protein) in 20 mM phosphate buffer at pH 7.0 for 2 h and heating the protein dispersion at 90 °C for 60 min. After cooling the hydrogels to room temperature to stop the crosslinking reaction, they were stored overnight at 4 °C. The protein hydrogels were mixed with phosphate buffer (1:1 w/w) and homogenized using a kitchen blender (HB711M, Kenwood, UK) for 5 min at speed 3. A vacuum oven was used to defoam the aqueous dispersion. Subsequently, the macroscopic dispersion of hydrogel particles was passed through a two-stage valve homogenizer (Panda, GEA Niro Soavi Homogeneizador Parma, Italy) two times at a pressure of 250/ 50 bar. The resulting aqueous dispersion of PPM contained 6.28 wt% pea protein. Sodium azide (0.02 wt%) was added as a bactericide. The PPM dispersion was further diluted to 1.25 or 3.33 wt% protein levels before its use in emulsion preparation.
Preparation of O/W Pickering Emulsions (PPM-E)
Pea protein microgel-stabilized Pickering emulsions (PPM-E) were fabricated by mixing sunflower oil and an aqueous dispersion of PPM (20: 80 w/w), where the PPM dispersion contained 1.25 wt% protein, at pH 7.0. The oil phase and the aqueous dispersion containing PPM were pre-sheared at 8,000 rpm using a rotor–stator type mixer (Silverson Shear Mixer, L5M-A, UK) for 5 min. To create fine emulsions, these pre-homogenized coarse emulsions were then passed through the Panda homogenizer at 250/ 50 bar pressure two times. For preparation of particle–particle-stabilized emulsions containing CNC, the CNC dispersions were prepared in Milli-Q water at pH 3.0 and stirred overnight. PPM-E + CNC were prepared in a similar way but oil: aqueous phase (40:60 w/w) was altered with the PPM dispersion containing 3.33 wt% protein. Subsequently, the 40.0 wt% O/W emulsion was adjusted to pH 3.0 and diluted with a dispersion of 2.0 or 6.0 wt% CNC particles and stirred for 3 h. In this way, all the emulsions prepared had the same 20.0 wt% oil and 1.0 wt% PPM as above but with + 1.0 wt% CNC (referred to as PPM-E + CNC1.0) or 3.0 wt% CNC (referred to as PPM-E + CNC3.0).
For the measurement of free fatty acid (FFA) release using the pH stat-based titrimetric method, a whey protein isolate (WPI)-stabilized emulsion was prepared and served as a control, using same methodology as for the PPM-E, but using 1.0 wt% WPI at pH 7.0. In addition, heat treated emulsions, i.e., PPM-E and PPM-E + CNC1.0 were heated at 90 °C for 30 min for FFA release measurements but without a prior gastric digestion step.
In vitro Gastrointestinal Digestion
In vitro gastrointestinal digestion based on the static INFOGEST model developed by Minekus and coworkers [33] was employed, but without the oral step since the emulsions did not contain any starch. Emulsion samples were incubated in simulated gastric fluid as previously reported by Zhang, Murray, Suriyachay, Holmes, Ettelaie and Sarkar [22]. Briefly, 10 mL of the emulsion sample at pH 3.0 was mixed with 10 mL of simulated gastric fluid (SGF) containing 2000 U mL−1 pepsin at pH 3.0 [33]. The mixture was incubated for 2 h at 37 °C using a shaking water bath (100 rpm, Grant Instruments Ltd, Cambridge, UK). For sequential intestinal digestion, the sample + SGF mixture was adjusted to the intestinal pH (pH 6.8) with 1.0 M NaOH and mixed with 3.18 mL simulated intestinal fluid (SIF): the recipe contained 3.570 g L−1 of NaHCO3, 1.123 g L−1 of NaCl, 0.335 g L−1 of MgCl2(H2O)6, 0.254 g L−1 of KCl, 0.054 g L−1 of KH2PO4, and freshly prepared bile acid solution (10 mM in the final digestion mixture), 0.02 mL of 0.3 M CaCl2, and pancreatin solution (2000 U/mL based on lipase activity in the final volume). The emulsion + SGF + SIF mixture was further incubated at 37 °C for 2 h using a shaking water bath (100 rpm, Grant Instruments Ltd, Cambridge, UK).
To separate out the effects of lipolysis alone on the emulsions, in some experiments the gastric phase was bypassed and lipase from porcine pancreas Type II (activity 68 U mg−1) was added in SIF buffer instead of pancreatin. Aliquots were collected at 0, 30 and 120 min and droplet size and zeta-potential measured immediately. Data described as being collected at “0 min SIF” refer to the aliquot at 0 min, i.e., gastric-digested sample + SIF buffer without any pancreatic enzymes or bile salts being added. At the same time, to try to separate out the effects of the bile salts, a mixture of post-gastric digested sample and SIF buffer without any added pancreatic enzymes was formed and named as “0 min SIF + bile”.
Sizing of PPM and Emulsion Droplets
The mean hydrodynamic diameters (dH) of the PPM dispersions as a function of intestinal digestion time were measured using dynamic light scattering (DLS) at 25 °C using a Zetasizer Nano-ZS (Malvern Instruments, Malvern UK). The aliquots were diluted 100-times in SIF buffer. Refractive index and absorbance value of 1.52 and 0.001 was used for PPM, respectively, as mentioned by Zhang et al. [22].
To measure the droplet size distributions of the Pickering emulsions before and after gastrointestinal digestion, static light scattering (SLS) was used employing a Mastersizer 3000 (Malvern Instruments Ltd, Malvern, Worcestershire, UK). The mean droplet size was reported as d32 (surface mean diameter,\(\frac{\sum {n}_{i }{d}_{i}^{3}}{\sum {n}_{i }{d}_{i}^{2}}\)) and d43 (the volume mean diameter,\(\frac{\sum {n}_{i }{d}_{i}^{4}}{\sum {n}_{i }{d}_{i}^{3}}\)) where, ni is the number of droplets with a diameter, di. In some cases, d(90) is reported meaning that 90% of the total droplets are smaller than this size. Refractive indices of 1.46 and 1.33 were used for sunflower oil and the aqueous phase, respectively.
Zeta-potential
A Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK) was used to measure zeta-potentials (ζ-potentials) of the particles and the emulsions. The samples were diluted to 0.01 wt% particle concentration (or 0.008 wt% droplet concentration) using the appropriate buffer (phosphate buffer, SGF or SIF), and then added into a folded capillary cell (DTS1070 cell, Malvern Instruments Ltd., Worcestershire, UK) for measurement of the electrophoretic mobilities and calculation of the ζ-potentials.
Confocal Laser Scanning Microscopy and Visual Imaging
Evolution of microstructures of PPM-E and PPM-E + CNC1.0 or PPM-E + CNC3.0 as a function of in vitro gastrointestinal digestion was probed using a confocal microscope (Zeiss LSM 880, Carl Zeiss MicroImaging GmbH, Jena, Germany). Approximately, 5 mL of sample was stained using Nile Red (2% w/v dispersion in dimethyl sulfoxide) for the oil phase, Nile Blue (10% w/v dispersion in Milli-Q water) for the PPM and Calcofluor White (1 mg/mL dispersion in Milli-Q water) for CNC. Multiple channels of 410, 514, 633 nm were used simultaneously to excite Calcofluor White, Nile Red and Nile Blue, respectively. In order to reduce Brownian motion of the droplets, few drops of a thickener i.e. xanthan gum solution (1.0 wt%) were added to the stained samples. The samples were imaged using 63 × (oil immersion) objective lens.
Samples were also stored glass tubes or Petri dishes and photographed with a digital camera in order to record any oiling off, creaming, aggregation or sedimentation.
Kinetics of Free Fatty Acids (FFAs) Release
An automated pH–stat-based titration unit (TIM 856 titration manager, Titralab, Radiometer analytical, UK) was used to monitor the kinetics of release of free fatty acids (FFAs) from the emulsions during in vitro intestinal digestion with or without bypassing the gastric digestion stage. The pH of the emulsion was maintained at pH 6.8 for 2 h and any FFAs released were neutralized using NaOH (0.05 M). The volumes of NaOH utilized allowed calculation of the quantity of FFAs generated from the emulsions, assuming 2 mol of FFAs were released per mole of triacylglycerol (1) [12]:
where, VNaOH is the required volume (mL) of NaOH added to neutralize the FFAs, MNaOH is the molarity of sodium hydroxide (0.05 M for PPM-E and PPM-E + CNC1.0 and PPM-E + CNC3.0, and 0.1 M for the WPI-stabilized emulsion), MwLipid is the molar mass of sunflower oil (0.880 kg mol−1) and WLipid is the mass of the lipid in the initial emulsion (2 g). The FFA release from the emulsions was analyzed using a nonlinear regression model (Eqs. (2) and (3)), as described by Sarkar and coworkers [9, 12]:
where, Φt (%) is the total quantity of FFAs released at time (t) during the simuated intestinal digestion; Φmax (%) is the maximum quantity of FFA released; k (mol s−1 m−2) is the lipid-FFA conversion rate per unit surface area of the emulsion droplet; D is the diffusion coefficient of the enzyme (lipase); n is the molar concentration of the lipase in the SIF; ρ0 is the density of the sunflower oil (910 kg m−3); d0 is the initial volume mean diameter of the emulsions and Γmax is the maximum coverage of the droplet surface by the lipase. Meanwhile, t1/2 is the time required to achieve 50% of the Φmax (%), calculated from Eq. (3):
Thus, the FFA versus digestion time curve was characterized via the following quantitative terms with Φ10 (the quantity of FFA released in the first 10 min), Φmax, k and t1/2, by fitting the experimental data to Eq. (3).
Statistical Analyses
All measurements were repeated three times on samples prepared on three separate days. Results are reported as the means and standard deviations (n = 3 × 3). One-way (ANOVA) was used for statistical analysis and the significant difference between samples were considered when p < 0.05 using Tukey’s adjustment. Model parameters for the release of FFAs were determined by minimisation of summed residuals.
Results and Discussion
Colloidal Stability of PPM Dispersions during Simulated Gastrointestinal Digestion
The PPMs used in this study were soft spherical particles with dH ranging from 230 to 300 nm at pH 7.0. As reported previously [22, 27], the PPM particles had a negative charge at pH 7.0 (~ -40 mV) and served as Pickering stabilizer of O/W emulsions irrespective of the pH. Before analyzing the effects of gastrointestinal treatments on the behavior of PPM-E, the effects of exposure to SIF, with or without prior exposure to SGF, was first studied for the PPM dispersions alone.
Figure 1a shows that the dH of PPM was essentially unchanged at time 0 on adding SIF + bile salts at pH 7.0 but without pancreatin (p > 0.1). This agrees with previous results, illustrating the high electrostatic and steric repulsion between the microgel particles [27]. However, since pancreatin contains various proteases [32,33,34], not surprisingly dH was affected on adding pancreatin along with SIF. Figure 1a shows that dH significantly increased from 0.27 to 1.7 μm after 30 min of incubation, and further increased to ~ 3 μm after 2 h digestion time (p < 0.05). The increase in dH was accompanied with increased polydispersity index (PDI), indicating PPM aggregation, which also agreed with the visual appearance of aggregates and sediments (see in Fig. 1b). Presumably hydrolysis of the chains on the surface of the PPMs occurs first and this could lead to reduced steric stability. However, the ζ-potential of PPM (-36 mV at 0 min) also slightly decreased in the first 30 min of digestion time (p < 0.05) but then remained steady for longer times (p > 0.1), even though dH continued to increase. These changes in ζ-potential were probably complicated by the adsorption of anionic bile salts to the PPM [12, 34, 35] in addition to changes due to proteolysis of the surface chains of protein in the PPMs. The variation of dH on exposure to SGF then SIF is shown in Fig. 1c. It is known that protein is “completely” digested by pepsin after 2 h [22], so the initial spherical structure is expected to be partly disintegrated early on, though again the initial result seemed to be the formation of larger (> 1 μm) aggregates with a high PDI. Proteolysis of protein-based fragments produced by gastric treatment can be enhanced in the intestinal phase depending on what kinds of bile salts are present [36, 37]. It is worth noting that the increase of dH in Fig. 1c (SGF then SIF) was similar to that in Fig. 1a (SIF only) (p > 0.1), suggesting that further addition of intestinal proteases did not influence the PPM size and structure [38, 39]. This might be expected owing to the significantly smaller size of the digestive enzymes compared to the typical mesh size in the PPM network, which may hydrolyze the PPM already in the gastric phase and thus the intestinal enzymes might not further alter the size of the PPM [40].

Evolution of mean hydrodynamic diameter (■), polydispersity index (PDI) (□) after intestinal digestion without gastric step (a); ζ-potential (■) of PPM after in vitro intestinal digestion as a function of time (0, 30 and 120 min) (b); or evolution of mean hydrodynamic diameter (
 ), polydispersity index (PDI) (
), polydispersity index (PDI) (
 ) after intestinal digestion with gastric step (c). Insets in (b) show the visual images of aqueous dispersion of PPM during intestinal digestion. Digestion time of 0 min in (a) shows the original PPM + SIF + bile salt mixture without the addition of pancreatin at pH 6.8. Digestion time of 0 min in (c) represents the gastric-digested PPM + SIF + bile salt mixture without the addition of pancreatin at pH 6.8. Standard deviations are shown by error bars
) after intestinal digestion with gastric step (c). Insets in (b) show the visual images of aqueous dispersion of PPM during intestinal digestion. Digestion time of 0 min in (a) shows the original PPM + SIF + bile salt mixture without the addition of pancreatin at pH 6.8. Digestion time of 0 min in (c) represents the gastric-digested PPM + SIF + bile salt mixture without the addition of pancreatin at pH 6.8. Standard deviations are shown by error bars
Behavior of Pickering Emulsions during Simulated Gastrointestinal Digestion
Evolution of Size of Droplets
Figure 2 shows the gradual evolution in the particle size distribution (PSD) of the PPM-E during simulated in vitro gastrointestinal digestion. Table 1 shows the d43, d32 and d(90) values for the PPM-E ± CNC. For the PPM-E without CNC, the PSD initially had two peaks with the smaller one ranging between 0.1 and 1.0 μm, as reported in previous papers [22, 33]. We assume that this smaller peak represents free PPM in the continuous phase since it completely disappeared after 2 h gastric digestion. After the gastric phase and adjusting the pH of the PPM-E to 7.0 and diluting with SIF buffer (0 min SIF, Fig. 2a), there was no noticeable change in the PSD: d43 reduced from 50 to 40 μm (p < 0.05) [22] whilst d32 was unaffected (p > 0.05). On addition of bile salts (Fig. 2a), the main peak moved to lower particle sizes, with a significant decrease in d32 but a steady value of d43. Adsorption of the bile salts to the PPM-E and remaining PPM obviously leads to some rearrangement of the aggregates of PPM-E and PPM, no doubt due to the negative charge on some bile salt molecules.

Mean size distribution of gastric-digested Pickering oil-in-water emulsions stabilized by PPM containing 20 wt% oil (PPM-E) (a); PPM-E further coated by 1.0 wt% CNC (PPM-E + CNC1.0) (b) 3.0 wt% CNC (PPM-E + CNC3.0) (c) during in vitro intestinal digestion. Digestion time of 0 min SIF shows the gastric-digested emulsion + SIF mixture without the addition of pancreatin and bile salts at pH 7.0. Time 0 min SIF + bile represents the gastric-digested emulsion + SIF + bile salt mixture without the addition of pancreatin at pH 7.0
When pancreatin was added the PSD became multi-modal, with a second peak appearing in the range 500–5000 μm, whilst the main peak moved to a larger size range and decreased in height. In line with these changes, d43 significantly increased, by nearly five times, (p < 0.05) whilst d32 nearly halved in the first half-an-hour of the intestinal digestion (see Table 1). With increasing incubation time, the height of first peak in the size range of 1 to 100 μm significantly reduced whilst the area of the second peak in the larger size range increased, in agreement with the 4 × increase in both d43 and d32 (Table 1). These changes suggested extensive droplet coalescence as well as aggregation, due to the pancreatic lipases accessing the oil droplets as the PPM particles become increasingly degraded by the protease action. At this stage the PPM fragments are obviously too small to provide effective Pickering stabilization and/or their surface activity is significantly reduced such that they are completely displaced by other surface active species that allow lipase adsorption. Of course, even when the PPM remain at the interface at their maximum packing density, if they remain spherical then there will still be gaps between the particles far larger than the size of a lipase molecule. For a contact angle of 90° the maximum width of the gap is (\(\frac{\sqrt{3}-1){d}_{H}}{2})\)≈ 85 to 110 nm (where dH is 230 to 300 nm), whereas the radius of the pancreatic lipase/co-lipase complex is only 2.5 nm [12]. Thus, lipase molecules can easily diffuse through the gaps between non-digested PPM at the interface, unless the PPM flatten and/or fuse together.
The initial behaviour of the PPM-E + CNC1.0 emulsions is quite different compared in the absence of CNC (see Fig. 2b) but addition of 3.0 wt% CNC does not change the PSD markedly (Fig. 2c). There is no significant change in the PSD after incubation in SGF with pepsin for 2 h. It is known that CNC protects PPM-E from complete peptic digestion [22]. The partial PPM fragments therefore seem to cooperate with the CNC to form an effective stabilizing layer around the oil droplets, no doubt due to the opposite charges on CNC and PPM at pH 3.0. The PSDs of the gastric-digested PPM-E + CNC1.0 or PPM-E + CNC3.0 show a prominent peak for 1 to 100 μm-sized emulsion droplets that moves to larger sizes when diluted with SIF at pH 7.0 but before addition of pancreatin: d43 increased from 12 to 13 μm (p < 0.05) (see Table 1). This suggests some droplet flocculation and/or coalescence. In presence of bile salts, the PSD shows a third peak in a smaller size range whilst the height of main peak is slightly reduced and both d43 and d32 values are slightly decreased (p < 0.05) (see Table 1).This implies that the bile salts cause some re-dispersal of the flocs to smaller sizes. On addition of pancreatin d43 significantly increased to nearly 30 fold after 30 min, reflecting the shift in the main peak to high droplet sizes in the PSD. With increased incubation time, the smaller peak in the PSD moved to a slightly higher size range with negligible change in the larger peak in both PPM-E + CNC1.0 and PPM-E + CNC3.0, and the overall d43 and d32 remained unaltered.
The variations in PSD of PPM-E + CNC3.0 were similar to that of PPM-E + CNC1.0. As shown in Table 1, there was a marked increase in d43 of gastric-digested PPM-E + CNC3.0 after pH adjustment from pH 3.0 to pH 7.0 with SIF ions, whilst addition of bile salts reduced d43. After addition of pancreatin, d43 significantly increased to more than 350 μm. The similarity in these trends indicates that the concentration of CNC (at least between 1 and 3 wt.%) has little influence on droplet stability in the intestinal phase. In other words, although CNC is efficacious in aiding the stability of droplets in gastric phase, CNC does not offer promising protection against intestinal destabilization.
Microstructural Changes
CLSM images of PPM-E ± CNC after sequential in vitro gastrointestinal digestion are shown in Figs. 3, 4, 5. The PPM and their fragments (labeled with Nile blue) are represented in green, the oil (labeled by Nile red) is represented in red and the CNC (labeled by Calcofluor white) is represented by blue colour. At 0 min SIF, PPM-E droplets are clearly seen to be surrounded by a proteinaceous layer, suggesting a residue of gastric-digested PPM fragments still remains at the interface. On addition of bile salts, this proteinaceous layer seems to disappear from around most oil droplets, especially the larger ones (Fig. 3). This qualitative observation, coupled with an increased green (protein) signal in the continuous phase, is good evidence of displacement of the adsorbed PPM fragments by the more surface active bile salts. Furthermore, the emulsion droplets appeared to be slightly deflocculated, in agreement with the reduction in d43 as discussed above (Table 1).

Confocal micrographs, visual images and schematic representations of PPM-stabilized emulsion (PPM-E) after sequential in vitro gastrointestinal digestion. Green colour (Nile Blue stain) represents PPM; the red colour (Nile Red stain) represents the oil phase and the black background is air or water. Digestion time of 0 min SIF represents the gastric- digested emulsion + SIF mixture without the addition of pancreatin and bile salts at pH 7.0. Digestion time of 0 min SIF + bile represents the gastric-digested emulsion + SIF + bile salt mixture without the addition of pancreatin at pH 7.0. Scale bar = 20 μm

Confocal micrographs and visual images of PPM stabilized emulsion coated with 1.0 wt% CNC after sequential in vitro gastrointestinal digestion. Green colour (Nile Blue stain) represents PPM; the red colour (Nile Red stain) represents the oil phase; the blue colour (Calcufluor White stain) represents the CNC and the black background represents air or water. Digestion time of 0 min SIF represents the gastric-digested emulsion + SIF mixture without the addition of pancreatin and bile salts at pH 7.0. Digestion time of 0 min SIF + bile represents the gastric-digested emulsion + SIF + bile salt mixture without the addition of pancreatin at pH 7.0. Scale bar = 20 μm

Confocal micrographs and visual images of PPM stabilized emulsion coated with 3.0 wt% CNC after sequential in vitro gastrointestinal digestion. Green colour (Nile Blue stain) represents PPM; the red colour (Nile Red stain) represents the oil phase; the blue colour (Calcufluor White stain) represents the CNC and the black background represents air or water. Digestion time of 0 min SIF represents the gastric-digested emulsion + SIF mixture without the addition of pancreatin and bile salts at pH 7.0. Digestion time of 0 min SIF + bile represents the gastric-digested emulsion + SIF + bile salt mixture without the addition of pancreatin at pH 7.0. Scale bar = 20 μm
After addition of pancreatin, the separated cream phase was replaced by a clear oil layer, clearly indicating significant coalescence. This is consistent with the dramatic increase in d43 (Table 1) and illustrates the dramatic effect of the combined digestive action of pancreatic lipases plus proteases [34, 35, 41,42,43,44]. The FFAs and mono-acylglycerols (MAG) generated at the surfaces of the emulsion droplets via the action of lipases [43, 45] whilst surface active and present in sufficient quantities to displace the bile salts, are less effective in preventing droplet coalescence.
The CLSM observations of PPM-E + CNC1.0 (Fig. 4) were quite similar to those recorded with PPM-E in the absence of CNC – significant coalescence was the end result. Since the enhanced stabilization with PPM and CNC in the gastric phase is dependent on their opposite charges at low pH, this mechanism cannot operate when the pH is raised to 7.0, in the intestinal phase, because the two types of particle are both negative and will repel one another. The CNC particles are most likely released into the bulk continuous phase, as Fig. 4 suggests, and this CNC now in the bulk would appear to have little effect on the course of the emulsion digestion. Interestingly, although the measured d43 still increased dramatically (Table 1), a CNC network appeared to form in the bulk phase as shown in the confocal micrograph and in non-diluted samples stored in test tubes. This inhibited the formation of a cream layer, unlike that observed for PPM-E in the absence of CNC.
For PPM-E + CNC3.0 (Fig. 5), the CLSM images were essentially no different from those with PPM-E + CNC1.0, except that there was a stronger blue signal due to the higher concentration of CNC. Addition of cellulose has been shown to significantly inhibit lipase action on emulsions [18, 19, 25, 45,46,47,48,49]. Sarkar et al. [45] studied an O/W emulsion stabilized by a complex of whey protein isolate (WPI) and CNC. They found that the emulsion droplets became encapsulated in a strong, gel-like CNC network and formed “raspberry-like” flocs, resulting in emulsion microgel-like particle. Such encapsulation could inhibit direct access of bile salts or digestive enzymes to the droplets. On the other hand, Singh et al. [25] stabilized Pickering emulsions using chitosan particles as the emulsifier and alginate as a coating and showed significant inhibition of lipid digestion of the oil droplets. However, excess alginate in the continuous phase significantly enhanced the viscosity of emulsions, and this was one of the reasons for the inhibition of the lipid digestion. Although such network-like structure was also observed in this study in the confocal images, the strength of the network in the bulk phase might not be sufficient to prevent the access of bile salts and lipase due to the limited concentration of CNC used.
ζ- potential
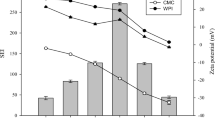
The ζ-potential values of emulsions at pH 7.0 and in presence of SIF without/with the addition of bile salts and/or pancreatin are shown in Fig. 6. The ζ-potential of PPM-E was nearly zero at pH 3.0 after gastric digestion [22]. After adjusting the pH to 7.0 but without SIF, the gastric digested PPM-E showed a rapid increase of negative charge, with ζ ~ –45 mV. The magnitude of the ζ-potential value reduced slightly (~ –33 mV) on addition of pancreatin only (p < 0.05) whilst the inclusion of bile salts enhanced the negative surface charge (~ –50 mV), as expected if the anionic surfactant components of bile become strongly adsorbed. In this respect, this agrees with the CLSM images (Fig. 3), where it seemed that the adsorbed protein material was displaced. Bile salts are known to be efficient displacers of a range of proteinaceous emulsifiers, for example, soy, caseins and whey protein [35, 45, 50], whilst particles are supposed to be harder to displace due to their high desorption energies [9]. However, as already discussed above, by the time the PPM-E reach the intestinal digestion stage, the adsorbed protein material will have already been degraded to some extent by the gastric pepsin-induced proteolysis, i.e. it is unlikely that intact PPM remain at the interface anyway. After the initial addition of both bile salts and pancreatin, the ζ-potential of the PPM-E droplets showed no significant change over the next 30 min, but by the end (2 h) of the SIF digestion stage ζ had increased in magnitude to –70 mV, presumably due to adsorption of lipolysis products including FFAs.

Mean ζ-potential of gastric-digested PPM-E, PPM-E + CNC1.0 and PPM-E + CNC3.0 at pH 7.0. Digestion time of 0 min SIF represents the gastric-digested emulsion + SIF mixture without the addition of pancreatin or bile salts at pH 7.0. Digestion time of SIF + bile represents the gastric- digested emulsion + SIF + bile salt mixture without the addition of pancreatin at pH 7.0. Standard deviations are represented by the error bars
PPM-E + CNC1.0 droplets after gastric digestion at pH 3 had ζ ~ –9 mV but after neutralization to pH 7.0 with SIF buffer ζ ~ –60 mV. In other words, the inclusion of 1% CNC seemed to have little effect on ζ. However, this was to be expected if the two adsorbing species are both negative at pH 7, in which case there is no reason why the CNC should remain at the interface. The magnitude of ζ reduced slightly to ~ –50 mV on the addition of pancreatin (p > 0.05) and slightly further increased to ~ –65 mV on addition of bile salts, again suggesting that bile salts displaced any other interfacial material. By 30 min, ζ-potential changed to –70 mV but then remained constant for the next 90 min, again agreeing with the suggestion from the PSDs (Fig. 2b) that droplet destabilisation of emulsion was completed within the first 30 min. The values of ζ-potential observed throughout the various digestion stages for PPM-E + CNC3.0 were very similar to those for PPM-E + CNC1.0 (Fig. 6), further emphasizing that CNC has little influence on the intestinal digestion of PPM-E after the gastric digestion step.
Lipid Digestion Kinetics
The release of FFAs from the emulsions, determined via the INFOGEST static model [32] is shown in Fig. 7. The corresponding parameters (FFA release at 10 min, Φ10, maximum quantity of FFA release, Φmax, lipid-FFA conversion rate constant, k and the time to achieve half of the Φmax, t1/2) are reported in Table 2. Since the pea protein concentrate used in this study to create the PPM had low solubility and was unsuitable as an emulsion stabilizer on its own [27], for comparison a conventional (i.e., non-microgel stabilized) emulsion was created using whey protein isolate (WPI) as the stabilizer. With the latter, the oil droplets are usually rapidly digested and FFAs and MAGs released due to the ease of access of the lipase to the interface [12, 45]. Thus, the WPI-stabilized emulsions generated Φmax ~ 69% (data not shown), consistent with the results of previous studies [51,52,53].

Release of free fatty acid during intestinal digestion (after prior-gastric digestion) of WPI-stabilized emulsion (black diamond), PPM-E (green circle), PPM-E + CNC1.0 (red square), and PPM-E + CNC3.0 (blue triangle). Error bars represent the standard deviations. The solid lines in each of the %FFA curves connecting the symbols (data points) are the best theoretical fits to the experimental data predicted using the mathematical model (Eq. (1)
In the case of PPM-E, the maximum FFA release of lipid digestion was significantly lower, as expected for Pickering emulsions, since the size of the droplets was two orders of magnitude higher (d43 ~ 25 to 40 μm) compared to the WPI-stabilized emulsions (d43 ~ 0.2 to 0.5 μm), hence the interfacial area much lower. The PPM-E stabilized emulsions after the gastric digestion step were rapidly hydrolysed in the first 10 min, generating Φ10 ~ 15%, followed by a slower release of FFA, reaching ~ 20% at the end of digestion (2 h). This ‘self-inhibition’ might be due to long-chain FFAs produced from the sunflower oil accumulating at the interface and inhibiting of further lipase activity [12, 54, 55]. Similar results were reported in previously. For example Zhao et al. [16] reported 15 to 20% of FFA release in the soy protein nanoparticle-stabilized Pickering emulsions. These Pickering emulsions were produced via enzymatically hydrolyzed soy protein and gave a significantly lower extent and rate of FFA release than the non-hydrolysed soy protein as stabilizer. The maximum FFA release of lipolysis of Pickering emulsions stabilized by zein colloidal particles also had a similar value (ɸmax = 19.5%) [18] and the extent of FFA release from Pickering emulsions stabilized by WPI microgels and kafirin nanoparticle-stabilized emulsions was 40% after the gastrointestinal digestion [12, 17]. Conversely, Meshulam and Lesmes [14] concluded that the FFA release from Pickering emulsions stabilized by lactoferrin nanoparticles or native lactoferrin was essentially the same, giving ‘final’ release values of 65%. In all these different digestion studies one should remember that different digestion protocols and conditions have been used, so that it may be misleading to compare absolute values of FFA release.
For PPM-E + CNC1.0, Φ10 was ~ 22%, significantly higher than that of PPM-E. Meanwhile, the rate of lipolysis was slightly lower, and t1/2 in PPM-E + CNC1.0 shorter (approximately half) compared to that required for PPM-E, as shown by the data in Table 2. The behaviour of PPM-E + CNC3.0 and PPM-E + CNC1.0 in terms of k and t1/2 were not significantly different (p \(>\) 0.05) but the Φmax of PPM-E + CNC3.0 was slightly higher and Φ10 was highest for PPM-E + CNC3.0. The increase in FFA release in PPM-E + CNC1.0–3.0 might be related to the larger surface area of the oil droplets generated after gastric digestion in presence of cellulose, due to secondary surface coverage [22], as compared with the coalesced PPM-E droplets after gastric digestion in the absence of CNC. However, it seems that there may still be some coverage of the droplets by CNC in the PPM-E + CNC1.0–3.0 samples, which reduces the rate of lipid digestion compared to in the absence of CNC (p < 0.05) (Table 2).
Lipid Digestion Kinetics Bypassing the Gastric Phase and Using Pure Lipase
Figure 8 shows the variation of the FFA release from PPM-E and PPM-E + CNC1.0–3.0 with and without the prior gastric digestion stage and without any intestinal proteolytic enzyme (trypsin). These experiments were also performed using the pure lipase from porcine pancreas Type II and bile salts. For PPM-E, Φ10 was significantly lower (almost half - 7.4% - see Table 3) compared to Φ10 with gastric proteolysis (15% - see Table 2), whilst Φmax ~ 13% and k ~ 0.63 μmol s−1 m−2. The value of t1/2 (8.4 min) was nearly 2 times longer when the prior gastric stage was excluded, suggesting that the bile salts have more difficulty in displacing the intact PPM from the interface compared to when they have undergone some proteolysis on the gastric stage. In the case of PPM-E + CNC1.0, Φmax was slightly lower (8%) compared to that of PPM-E, while k was similar (0.72 μmol s−1 m−2- see Table 3). Similar results were reported in previous work, i.e., a slight reduction in Φmax with addition of soluble polysaccharides, such as chitosan, pectin, methyl-cellulose, propylene glycol alginate or insoluble chitin nanocrystals [19, 46, 56, 57].

Release of free fatty acids (FFA%) during in vitro intestinal digestion bypassing the gastric phase (using pure lipase) with the inset zooming in on the time range 0 to 60 min of PPM-E (green circle), PPM-E + CNC1.0 (red square), and PPM-E + CNC3.0 (blue triangle). Error bars represent standard deviations. Solid lines connecting the symbols (data points) in each of the %FFA curves are the best theoretical fits to the experimental data predicted using the mathematical model (Eq. 1)
The t1/2 of PPM-E + CNC1 was almost three times shorter than that of PPM-E, also suggesting more rapid accumulation of lipid hydrolysis products at the O/W interface that inhibit further lipase activity. It is interesting to note that the post-gastric digestion step had no influence on the rate constant k or t1/2, whilst Φmax was 3 × lower than when the gastric phase was excluded, again highlighting the importance of proteolysis in the overall lipid digestion process (compare Tables 2 and 3). At the higher concentration of CNC, i.e., PPM-E + CNC3.0, Table 3 shows that k was lower (0.31 μmol s−1 m−2) compared to PPM-E + CNC1.0, whilst t1/2 was nearly 2 × longer and Φmax was similar. The reason for the similar extent but reduced rate of lipolysis of PPM-E + CNC1.0 and PPM-E + CNC3.0 might be a difference in the viscosities of the emulsions – higher viscosity would be expected to slow down the rate of mass transport of enzyme to the interface and the products from the interface [46]. PPM-E + CNC3.0 has a viscosity 100 × higher than PPM-E + CNC1.0 [22], although in this work the emulsions were diluted by a factor of 2 into the SIF buffer (as described in the Methods section) and small molecules like bile salts and lipase would be expected to easily pass through the pores in the network of CNC crystals [46].
Lipid Digestion Kinetics of Heat-treated Emulsions
One way of possibly improving the barrier properties of an adsorbed microgel layer is to heat the system above the melting point of the gel network, so that the individual particles might fuse to form a more coherent layer. At the same time, heating is relevant to practical applications, since most emulsions for consumer use would probably also undergo a pasteurization step. Therefore, samples of PPM-E and PPM-E + CNC1.0 (without a prior gastric stage) were heat treated at 90 ℃ at pH 7.0 for 30 min and their subsequent in vitro intestinal stage digestion was measured as previously. The results are shown in Fig. 9. It is interesting to note that HT-PPM-E had a significant lower Φmax than the unheated PPM-E (Fig. 9a). Possibly this indicates that some PPM fusion had occurred as hypothesized [12]. In contrast, there was no significant difference between PPM-E + CNC1.0 and HT-PPM-E + CNC1.0 in the overall kinetics of FFA (Fig. 9b), suggesting that the heat treatment did not the improve barrier properties of the mixed PPM + CNC adsorbed film, although it is again noted that at pH 7.0 the two types of particle will repel each other electrostatically.

Release of free fatty acids (%FFA) during in vitro intestinal digestion bypassing the gastric phase (using pure lipase with no added proteases) over time of (a) PPM-E and (b) PPM-E + CNC1.0 with (open circle) or without heat treatment (solid circle). Error bars represent standard deviations
Conclusions
Pickering emulsions stabilized by pea protein microgel (PPM) particles with the addition of cellulose nanocrystals (CNC) were tested to understand the influence of complex particle systems in modulating lipid digestion. The CNC were shown to inhibit the coalescence of oil droplets in the in vitro gastric stage, probably due to electrostatic attraction between the anionic CNC and the cationic PPM at the interface at low pH. Even though this attractive charge interaction should not be present during the intestinal digestion stage at pH 7.0 (where the particles are both negatively-charged), unprecedented results from this study reveal that in the presence of CNC lipolysis was slightly delayed, but the overall extent (at least after 2 h) of lipid digestion increased. To our knowledge, such an increase in FFA release is not observed in any other particle–particle interface study to date. These novel results suggests that such ‘dual particle’ Pickering emulsions, i.e., with a mixed interfacial particle layer, could be used to protect lipophilic bioactive compounds in the stomach by creating more stable droplets under gastric conditions, without inhibiting their release them in the intestines.
References
D.J. McClements, E.A. Decker, J. Weiss, J. Food. Sci. 72, R109 (2007)
H. Singh, A. Ye, D. Horne, Prog. Lipid. Res. 48, 92 (2009)
D.J. McClements, Annu. Rev. Food. Sci. Technol. 1, 241 (2010)
R.C. Benshitrit, C.S. Levi, S.L. Tal, E. Shimoni, U. Lesmes, Food. Funct. 3, 10 (2012)
K.P. Velikov, E. Pelan, Soft. Matter. 4, 1964 (2008)
B.P. Binks, Curr. Opin. Colloid Interface Sci. 7, 21 (2002)
B.P. Binks, T.S. Horozov, in Colloidal Particles at Liquid Interfaces. ed. by B.P. Binks, T.S. Horozov (Cambridge University Press, Cambridge, 2006), p. 1
E. Dickinson, Curr. Opin. Colloid Interface Sci. 15, 40 (2010)
A. Sarkar, S. Zhang, M. Holmes, R. Ettelaie, Adv. Colloid. Interface. Sci. 263, 195 (2019)
Q. Guo, A. Ye, N. Bellissimo, H. Singh, D. Rousseau, Prog. Lipid Res. 68, 109 (2017)
A. Sarkar, E. Dickinson, Curr. Opin. Colloid Interface Sci. 49, 69 (2020)
A. Sarkar, B. Murray, M. Holmes, R. Ettelaie, A. Abdalla, X. Yang, Soft. Matter. 12, 3558 (2016)
G. Shimoni, C. Shani Levi, S. Levi Tal, U. Lesmes, Food Hydrocolloids 33, 264 (2013)
D. Meshulam, U. Lesmes, Food Funct. 5, 65 (2014)
F. Liu, C.-H. Tang, Food Hydrocolloids 60, 631 (2016)
M. Zhao, P. Shen, Y. Zhang, M. Zhong, Q. Zhao, F. Zhou, ACS Food Sci. Technol. 1, 193 (2021)
J. Xiao, C. Li, Q. Huang, J. Agric. Food Chem. 63, 10263 (2015)
Y. Wei, Z. Liu, A. Guo, A. Mackie, L. Zhang, W. Liao, L. Mao, F. Yuan, Y. Gao, J. Agric. Food Chem. 69, 12278 (2021)
Y. Wei, D. Zhou, A. Mackie, S. Yang, L. Dai, L. Zhang, L. Mao, Y. Gao, J Agric. Food Chem. 69, 1619 (2021)
F. Ning, X. Wang, H. Zheng, K. Zhang, C. Bai, H. Peng, Q. Huang, H. Xiong, J. Funct. Foods 55, 76 (2019)
Y. Shao, C.-H. Tang, Food Res. Int. 79, 64 (2016)
B. Zhang, S. Murray, N. Suriyachay, M. Holmes, R. Ettelaie, A. Sarkar, Langmuir 37, 827 (2021)
F.A. Bellesi, M.J. Martinez, V.M. Pizones Ruiz-Henestrosa, A.M.R. Pilosof, Food Hydrocolloids 52, 47 (2016)
Y. Wei, Z. Tong, L. Dai, D. Wang, P. Lv, J. Liu, L. Mao, F. Yuan, Y. Gao, Food Hydrocolloids 104 (2020)
C.K. Surjit Singh, H.P. Lim, B.T. Tey, E.S. Chan, Carbohydr. Polym. 251, 117110 (2021)
A. Araiza-Calahorra, Y. Wang, C. Boesch, Y. Zhao, A. Sarkar, Curr. Res. Food Sci. 3, 178 (2020)
S. Zhang, M. Holmes, R. Ettelaie, A. Sarkar, Food Hydrocolloids 102, 105583 (2020)
S. Beck-Candanedo, M. Roman, D.G. Gray, Biomacromol. 6, 1048 (2005)
J. George, S.N. Sabapathi, Nanotechnol. Sci. Appl. 8, 45 (2015)
M. Younas, A. Noreen, A. Sharif, A. Majeed, A. Hassan, S. Tabasum, A. Mohammadi, K.M. Zia, Int. J. Biol. Macromol. 124, 591 (2019)
R. Moon, S. Beck, and A. Rudie, Production and applications of cellulose nanomaterials. 9 (2013)
A. Brodkorb et al., Nat. Protoc. 14, 991 (2019)
M. Minekus et al., Food Funct. 5, 1113 (2014)
S. Mun, E.A. Decker, D.J. McClements, Food Res. Int. 40, 770 (2007)
A. Sarkar, A. Ye, H. Singh, Food Hydrocolloids 60, 77 (2016)
J. Gass, H. Vora, A.F. Hofmann, G.M. Gray, C. Khosla, Gastroenterology 133, 16 (2007)
G. Martos, P. Contreras, E. Molina, R. Lopez-Fandino, J. Agric. Food Chem. 58, 5640 (2010)
H. Bokkhim, N. Bansal, L. Grøndahl, B. Bhandari, Food Hydrocolloids 52, 231 (2016)
Z. Zhang, R. Zhang, D.J. McClements, Food. Res. Int. 100, 86 (2017)
A. Sarkar, J.M. Juan, E. Kolodziejczyk, S. Acquistapace, L. Donato-Capel, T.J. Wooster, J. Agric. Food Chem. 63, 8829 (2015)
A. Haque, E.R. Morris, Carbohyd. Polym. 22, 161 (1993)
J. Maldonado-Valderrama, N.C. Woodward, A.P. Gunning, M.J. Ridout, F.A. Husband, A.R. Mackie, V.J. Morris, P.J. Wilde, Langmuir 24, 6759 (2008)
A. Sarkar, D.S. Horne, H. Singh, Int. Dairy J. 20, 589 (2010)
P. Wilde, B. Chu, Adv. Coll. Interface. Sci. 165, 14 (2011)
A. Sarkar, H. Li, D. Cray, S. Boxall, Food Hydrocolloids 77, 436 (2018)
M. Espinal-Ruiz, F. Parada-Alfonso, L.P. Restrepo-Sanchez, C.E. Narvaez-Cuenca, D.J. McClements, Food Funct. 5, 3083 (2014)
L. Bai, S. Huan, W. Xiang, L. Liu, Y. Yang, R.W.N. Nugroho, Y. Fan, O.J. Rojas, ACS Sustain. Chem. Eng. 7, 6497 (2019)
R. Meng, Z. Wu, Q.-T. Xie, B. Zhang, X.-L. Li, W.-J. Liu, H. Tao, P.-J. Li, Food Hydrocolloids 108, 106020 (2020)
M. Ding, L. Liu, T. Zhang, N. Tao, X. Wang, J. Zhong, Food Chem. 336, 127686 (2021)
T.T.P. Nguyen, B. Bhandari, J. Cichero, S. Prakash, Food Res. Int. 76, 348 (2015)
C. E. Sandoval-Cuellar, M. de Jesus Perea-Flores, and M. X. Quintanilla-Carvajal, J. Food Eng. 278 (2020)
A. Ye, X. Wang, Q. Lin, J. Han, H. Singh, Food Chem. 318, 126463 (2020)
R. Zhang, Z. Zhang, H. Zhang, E.A. Decker, D.J. McClements, Food Res. Int. 75, 71 (2015)
M.V. Tzoumaki, T. Moschakis, E. Scholten, C.G. Biliaderis, Food Funct. 4, 121 (2013)
Y. Li, D.J. McClements, J. Agric. Food Chem. 58, 8085 (2010)
M. Jo, C. Ban, K.K.T. Goh, Y.J. Choi, Food Hydrocolloids 94, 603 (2019)
H. Zhou, T. Dai, J. Liu, Y. Tan, L. Bai, O.J. Rojas, D.J. McClements, Food Hydrocolloids 113, 106494 (2021)
Author information
Authors and Affiliations
Contributions
SZ performed the experiments, prepared the figures and tables, analyzed the data regarding and wrote the manuscript. AS designed the experimental protocol. MH contributed to theoretical fitting of the digestion data. BSM, RE, MH and AS contributed to data interpretation and editing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Murray, B.S., Holmes, M. et al. Gastrointestinal Fate and Fatty Acid Release of Pickering Emulsions Stabilized by Mixtures of Plant Protein Microgels + Cellulose Particles: an In Vitro Static Digestion Study. Food Biophysics 18, 120–132 (2023). https://doi.org/10.1007/s11483-022-09756-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11483-022-09756-5