IgE Antibodies against Cancer: Efficacy and Safety

 ,
,  and
and 
Abstract
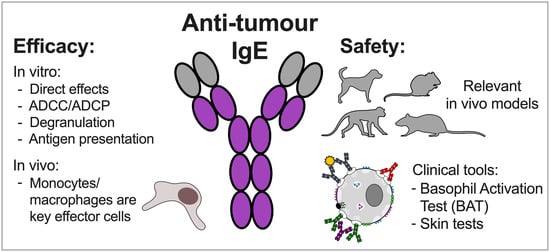
:
1. Introduction
2. Pre-Clinical Efficacy of Antitumour IgE Antibodies
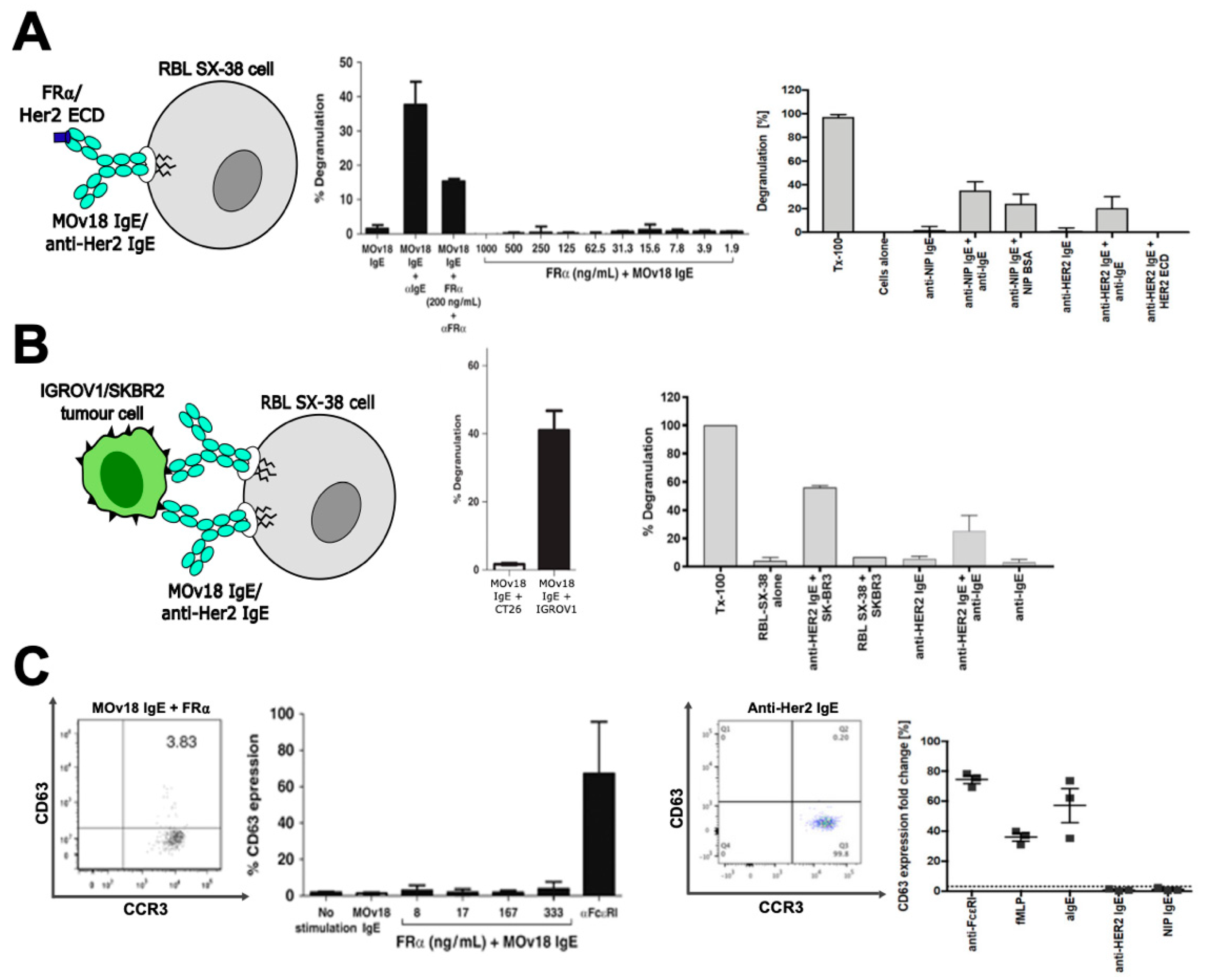
2.1. In Vitro Studies
2.2. In Vivo Studies
2.3. Monocytes and Macrophages are Key Effector Cells in the Mechanism of Action of Anti-Cancer IgE
3. Considering the Safety of Anti-Cancer IgE Therapeutics
3.1. IgE and Type I Hypersensitivity
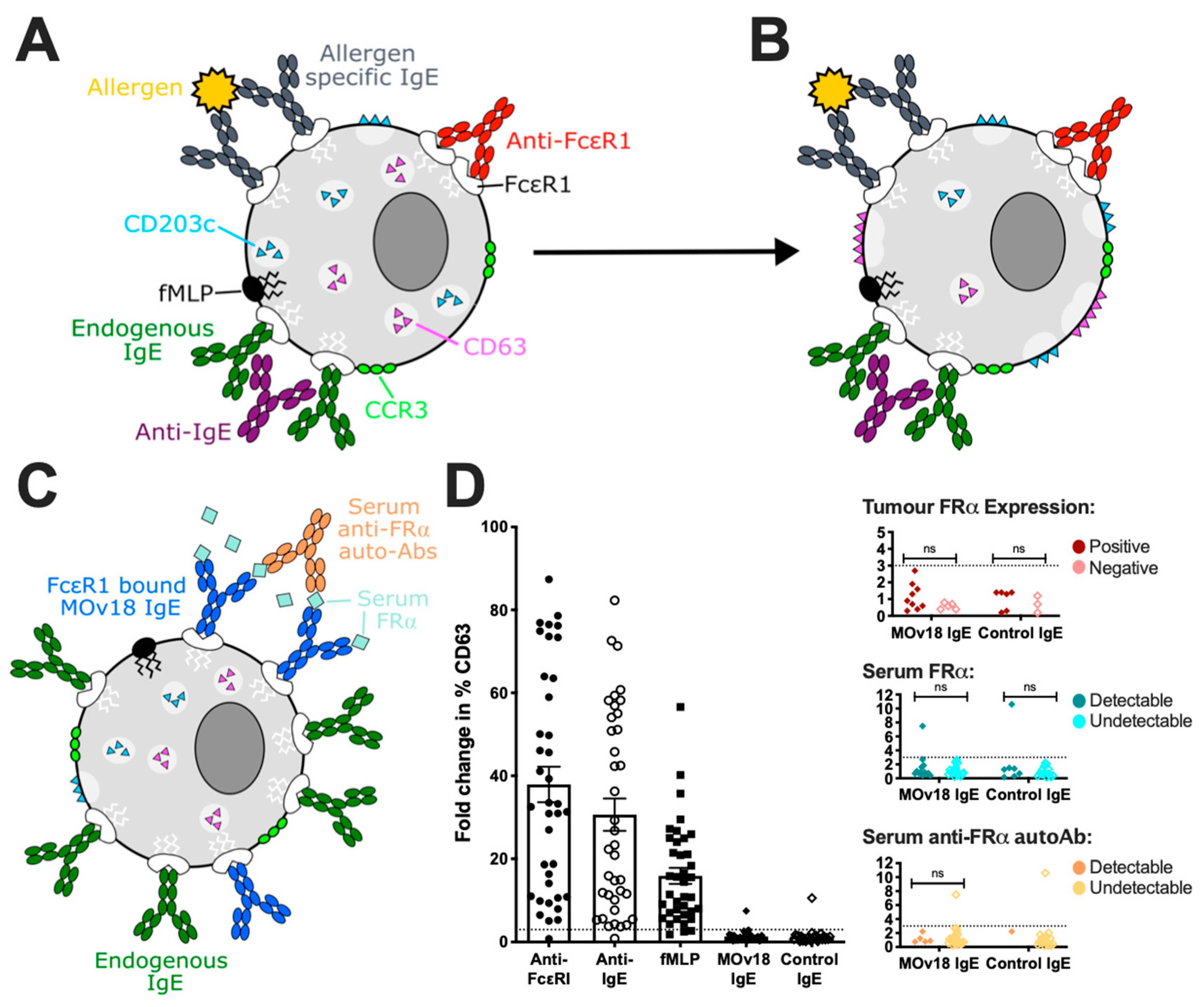
3.2. In Vitro Studies of Potential Hypersensitivity Reactions to Therapeutic IgE
3.3. Pre-Clinical In Vivo Safety Studies of Anticancer IgE Therapeutics
3.3.1. Cynomolgus Monkey
3.3.2. Canine
3.3.3. Rat
3.4. IgE Antibodies Are Heavily Glycosylated
3.5. Clinical Tools to Predict and Monitor Safety
3.5.1. Skin Tests
3.5.2. Basophil Activation Test (BAT)
3.5.3. First-in-Human Study of MOv18 IgE
4. Future Development of IgE Immunotherapy
Author Contributions
Funding
Conflicts of Interest
References
- Wulaningsih, W.; Holmberg, L.; Garmo, H.; Karagiannis, S.N.; Ahlstedt, S.; Malmstrom, H.; Lambe, M.; Hammar, N.; Walldius, G.; Jungner, I.; et al. Investigating the association between allergen-specific immunoglobulin E, cancer risk and survival. Oncoimmunology 2016, 5, e1154250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Hill, A.W. Atopy and Specific Cancer Sites: A Review of Epidemiological Studies. Clin. Rev. Allergy Immunol. 2016, 51, 338–352. [Google Scholar] [CrossRef]
- Schwartzbaum, J.; Seweryn, M.; Holloman, C.; Harris, R.; Handelman, S.K.; Rempala, G.A.; Huang, R.P.; Burkholder, B.; Brandemihl, A.; Kallberg, H.; et al. Association between Prediagnostic Allergy-Related Serum Cytokines and Glioma. PLoS ONE 2015, 10, e0137503. [Google Scholar] [CrossRef] [PubMed]
- Josephs, D.H.; Spicer, J.F.; Corrigan, C.J.; Gould, H.J.; Karagiannis, S.N. Epidemiological associations of allergy, IgE and cancer. Clin. Exp. Allergy 2013, 43, 1110–1123. [Google Scholar] [CrossRef]
- Ferastraoaru, D.; Rosenstreich, D. IgE deficiency is associated with high rates of new malignancies: Results of a longitudinal cohort study. J. Allergy Clin. Immunol. Pract. 2020, 8, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Neuchrist, C.; Kornfehl, J.; Grasl, M.; Lassmann, H.; Kraft, D.; Ehrenberger, K.; Scheiner, O. Distribution of immunoglobulins in squamous cell carcinoma of the head and neck. Int. Arch. Allergy Immunol. 1994, 104, 97–100. [Google Scholar] [CrossRef]
- Fu, S.L.; Pierre, J.; Smith-Norowitz, T.A.; Hagler, M.; Bowne, W.; Pincus, M.R.; Mueller, C.M.; Zenilman, M.E.; Bluth, M.H. Immunoglobulin E antibodies from pancreatic cancer patients mediate antibody-dependent cell-mediated cytotoxicity against pancreatic cancer cells. Clin. Exp. Immunol. 2008, 153, 401–409. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, D.; Cai, S.; Li, Q.; Li, X. Circulating basophil count as a prognostic marker of tumor aggressiveness and survival outcomes in colorectal cancer. Clin. Transl. Med. 2020, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zhang, X.; Wang, G.; Zhou, Y.; Luo, M.; Wang, S.; Hong, C. The impacts of pretreatment circulating eosinophils and basophils on prognosis of stage-colorectal cancer. Asia Pac. J. Clin. Oncol. 2018, 14, e243–e251. [Google Scholar] [CrossRef]
- Bax, H.J.; Chauhan, J.; Stavraka, C.; Khiabany, A.; Nakamura, M.; Pellizzari, G.; Ilieva, K.M.; Lombardi, S.; Gould, H.J.; Corrigan, C.J.; et al. Basophils from Cancer Patients Respond to Immune Stimuli and Predict Clinical Outcome. Cells 2020, 9, 1631. [Google Scholar] [CrossRef] [PubMed]
- Crawford, G.; Hayes, M.D.; Seoane, R.C.; Ward, S.; Dalessandri, T.; Lai, C.; Healy, E.; Kipling, D.; Proby, C.; Moyes, C.; et al. Epithelial damage and tissue gammadelta T cells promote a unique tumor-protective IgE response. Nat. Immunol. 2018, 19, 859–870. [Google Scholar] [CrossRef]
- Hayes, M.D.; Ward, S.; Crawford, G.; Seoane, R.C.; Jackson, W.D.; Kipling, D.; Voehringer, D.; Dunn-Walters, D.; Strid, J. Inflammation-induced IgE promotes epithelial hyperplasia and tumour growth. Elife 2020, 9. [Google Scholar] [CrossRef]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J.; Sutton, B.J.; Beavil, A.J.; Beavil, R.L.; McCloskey, N.; Coker, H.A.; Fear, D.; Smurthwaite, L. The biology of IGE and the basis of allergic disease. Annu. Rev. Immunol. 2003, 21, 579–628. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Zimel, M.; Heath, E.; Hogan, V.; Raz, A. Galectin-3 in bone tumor microenvironment: A beacon for individual skeletal metastasis management. Cancer Metastasis Rev. 2016, 35, 333–346. [Google Scholar] [CrossRef]
- Nakajima, K.; Kho, D.H.; Yanagawa, T.; Harazono, Y.; Hogan, V.; Chen, W.; Ali-Fehmi, R.; Mehra, R.; Raz, A. Galectin-3 Cleavage Alters Bone Remodeling: Different Outcomes in Breast and Prostate Cancer Skeletal Metastasis. Cancer Res. 2016, 76, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, T.; Morishita, A.; Fujihara, S.; Iwama, H.; Niki, T.; Fujita, K.; Akashi, E.; Mimura, S.; Oura, K.; Sakamoto, T.; et al. Galectin-9: An anticancer molecule for gallbladder carcinoma. Int. J. Oncol. 2016, 48, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Takano, J.; Morishita, A.; Fujihara, S.; Iwama, H.; Kokado, F.; Fujikawa, K.; Fujita, K.; Chiyo, T.; Tadokoro, T.; Sakamoto, T.; et al. Galectin-9 suppresses the proliferation of gastric cancer cells in vitro. Oncol. Rep. 2016, 35, 851–860. [Google Scholar] [CrossRef]
- Dreskin, S.C.; Goldsmith, P.K.; Strober, W.; Zech, L.A.; Gallin, J.I. Metabolism of immunoglobulin E in patients with markedly elevated serum immunoglobulin E levels. J. Clin. Investig. 1987, 79, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Iio, A.; Waldmann, T.A.; Strober, W. Metabolic study of human IgE: Evidence for an extravascular catabolic pathway. J. Immunol. 1978, 120, 1696–1701. [Google Scholar] [PubMed]
- Jones, E.A.; Waldmann, T.A. The mechanism of intestinal uptake and transcellular transport of IgG in the neonatal rat. J. Clin. Investig. 1972, 51, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Woodfolk, J.A.; Schuyler, A.J.; Stillman, L.C.; Chapman, M.D.; Platts-Mills, T.A. Half-life of IgE in serum and skin: Consequences for anti-IgE therapy in patients with allergic disease. J. Allergy Clin. Immunol. 2017, 139, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Jarolim, E.; Bax, H.J.; Bianchini, R.; Capron, M.; Corrigan, C.; Castells, M.; Dombrowicz, D.; Daniels-Wells, T.R.; Fazekas, J.; Fiebiger, E.; et al. AllergoOncology-the impact of allergy in oncology: EAACI position paper. Allergy 2017, 72, 866–887. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Jarolim, E.; Bax, H.J.; Bianchini, R.; Crescioli, S.; Daniels-Wells, T.R.; Dombrowicz, D.; Fiebiger, E.; Gould, H.J.; Irshad, S.; Janda, J.; et al. AllergoOncology: Opposite outcomes of immune tolerance in allergy and cancer. Allergy 2018, 73, 328–340. [Google Scholar] [CrossRef]
- Sutton, B.J.; Davies, A.M.; Bax, H.J.; Karagiannis, S.N. IgE Antibodies: From Structure to Function and Clinical Translation. Antibodies (Basel) 2019, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Singer, J.; Jensen-Jarolim, E. IgE-based immunotherapy of cancer: Challenges and chances. Allergy 2014, 69, 137–149. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Wang, Q.; East, N.; Burke, F.; Riffard, S.; Bracher, M.G.; Thompson, R.G.; Durham, S.R.; Schwartz, L.B.; Balkwill, F.R.; et al. Activity of human monocytes in IgE antibody-dependent surveillance and killing of ovarian tumor cells. Eur. J. Immunol. 2003, 33, 1030–1040. [Google Scholar] [CrossRef]
- Karagiannis, S.N.; Bracher, M.G.; Beavil, R.L.; Beavil, A.J.; Hunt, J.; McCloskey, N.; Thompson, R.G.; East, N.; Burke, F.; Sutton, B.J.; et al. Role of IgE receptors in IgE antibody-dependent cytotoxicity and phagocytosis of ovarian tumor cells by human monocytic cells. Cancer Immunol. Immunother. 2008, 57, 247–263. [Google Scholar] [CrossRef]
- Gould, H.J.; Mackay, G.A.; Karagiannis, S.N.; O’Toole, C.M.; Marsh, P.J.; Daniel, B.E.; Coney, L.R.; Zurawski, V.R., Jr.; Joseph, M.; Capron, M.; et al. Comparison of IgE and IgG antibody-dependent cytotoxicity in vitro and in a SCID mouse xenograft model of ovarian carcinoma. Eur. J. Immunol. 1999, 29, 3527–3537. [Google Scholar] [CrossRef]
- Josephs, D.H.; Bax, H.J.; Dodev, T.; Georgouli, M.; Nakamura, M.; Pellizzari, G.; Saul, L.; Karagiannis, P.; Cheung, A.; Herraiz, C.; et al. Anti-Folate Receptor-alpha IgE but not IgG Recruits Macrophages to Attack Tumors via TNFalpha/MCP-1 Signaling. Cancer Res. 2017, 77, 1127–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platzer, B.; Elpek, K.G.; Cremasco, V.; Baker, K.; Stout, M.M.; Schultz, C.; Dehlink, E.; Shade, K.C.; Anthony, R.M.; Blumberg, R.S.; et al. IgE/FcεRI-Mediated Antigen Cross-Presentation by Dendritic Cells Enhances Anti-Tumor Immune Responses. Cell Rep. 2015, 10, 1487–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen-Jarolim, E.; Achatz, G.; Turner, M.C.; Karagiannis, S.; Legrand, F.; Capron, M.; Penichet, M.L.; Rodriguez, J.A.; Siccardi, A.G.; Vangelista, L.; et al. AllergoOncology: The role of IgE-mediated allergy in cancer. Allergy 2008, 63, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Dodev, T.S.; Karagiannis, P.; Gilbert, A.E.; Josephs, D.H.; Bowen, H.; James, L.K.; Bax, H.J.; Beavil, R.; Pang, M.O.; Gould, H.J.; et al. A tool kit for rapid cloning and expression of recombinant antibodies. Sci. Rep. 2014, 4, 5885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, K.M.; Fazekas-Singer, J.; Bax, H.J.; Crescioli, S.; Montero-Morales, L.; Mele, S.; Sow, H.S.; Stavraka, C.; Josephs, D.H.; Spicer, J.F.; et al. AllergoOncology: Expression platform development and functional profiling of an anti-HER2 IgE antibody. Allergy 2019, 74, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Fazekas-Singer, J.; Singer, J.; Ilieva, K.M.; Matz, M.; Herrmann, I.; Spillner, E.; Karagiannis, S.N.; Jensen-Jarolim, E. AllergoOncology: Generating a canine anticancer IgE against the epidermal growth factor receptor. J. Allergy Clin. Immunol. 2018, 142, 973–976. [Google Scholar] [CrossRef]
- Crescioli, S.; Chiaruttini, G.; Mele, S.; Ilieva, K.M.; Pellizzari, G.; Spencer, D.I.R.; Gardner, R.A.; Lacy, K.E.; Spicer, J.F.; Tutt, A.N.J.; et al. Engineering and stable production of recombinant IgE for cancer immunotherapy and AllergoOncology. J. Allergy Clin. Immunol. 2018, 141, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Montero-Morales, L.; Maresch, D.; Crescioli, S.; Castilho, A.; Ilieva, K.M.; Mele, S.; Karagiannis, S.N.; Altmann, F.; Steinkellner, H. In Planta Glycan Engineering and Functional Activities of IgE Antibodies. Front. Bioeng. Biotechnol. 2019, 7, 242. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Turner, M.C.; Karagiannis, S.N. AllergoOncology: IgE- and IgG4-mediated immune mechanisms linking allergy with cancer and their translational implications. J. Allergy Clin. Immunol. 2017, 140, 982–984. [Google Scholar] [CrossRef] [Green Version]
- Ferastraoaru, D.; Bax, H.J.; Bergmann, C.; Capron, M.; Castells, M.; Dombrowicz, D.; Fiebiger, E.; Gould, H.J.; Hartmann, K.; Jappe, U.; et al. AllergoOncology: Ultra-low IgE, a potential novel biomarker in cancer—A Position Paper of the European Academy of Allergy and Clinical Immunology (EAACI). Clin. Transl. Allergy 2020, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, S.N.; Bracher, M.G.; Hunt, J.; McCloskey, N.; Beavil, R.L.; Beavil, A.J.; Fear, D.J.; Thompson, R.G.; East, N.; Burke, F.; et al. IgE-antibody-dependent immunotherapy of solid tumors: Cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J. Immunol. 2007, 179, 2832–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, P.; Singer, J.; Hunt, J.; Gan, S.K.; Rudman, S.M.; Mechtcheriakova, D.; Knittelfelder, R.; Daniels, T.R.; Hobson, P.S.; Beavil, A.J.; et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol. Immunother. 2009, 58, 915–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillner, E.; Plum, M.; Blank, S.; Miehe, M.; Singer, J.; Braren, I. Recombinant IgE antibody engineering to target EGFR. Cancer Immunol. Immunother. 2012, 61, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Z.; Utz, P.J.; Mollick, J.A. Using the allergic immune system to target cancer: Activity of IgE antibodies specific for human CD20 and MUC1. Cancer Immunol. Immunother. 2012, 61, 2295–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellizzari, G.; Hoskin, C.; Crescioli, S.; Mele, S.; Gotovina, J.; Chiaruttini, G.; Bianchini, R.; Ilieva, K.; Bax, H.J.; Papa, S.; et al. IgE re-programs alternatively-activated human macrophages towards pro-inflammatory anti-tumoural states. EBioMedicine 2019. [Google Scholar] [CrossRef] [Green Version]
- Daniels, T.R.; Leuchter, R.K.; Quintero, R.; Helguera, G.; Rodriguez, J.A.; Martinez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; Penichet, M.L. Targeting HER2/neu with a fully human IgE to harness the allergic reaction against cancer cells. Cancer Immunol. Immunother. 2012, 61, 991–1003. [Google Scholar] [CrossRef] [Green Version]
- Daniels-Wells, T.R.; Helguera, G.; Leuchter, R.K.; Quintero, R.; Kozman, M.; Rodriguez, J.A.; Ortiz-Sanchez, E.; Martinez-Maza, O.; Schultes, B.C.; Nicodemus, C.F.; et al. A novel IgE antibody targeting the prostate-specific antigen as a potential prostate cancer therapy. BMC Cancer 2013, 13, 195. [Google Scholar] [CrossRef] [Green Version]
- Rudman, S.M.; Josephs, D.H.; Cambrook, H.; Karagiannis, P.; Gilbert, A.E.; Dodev, T.; Hunt, J.; Koers, A.; Montes, A.; Taams, L.; et al. Harnessing engineered antibodies of the IgE class to combat malignancy: Initial assessment of FcvarepsilonRI-mediated basophil activation by a tumour-specific IgE antibody to evaluate the risk of type I hypersensitivity. Clin. Exp. Allergy 2011, 41, 1400–1413. [Google Scholar] [CrossRef]
- Nagy, E.; Berczi, I.; Sehon, A.H. Growth inhibition of murine mammary carcinoma by monoclonal IgE antibodies specific for the mammary tumor virus. Cancer Immunol. Immunother. 1991, 34, 63–69. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Darcy, P.K.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Tumor-specific IgE-mediated inhibition of human colorectal carcinoma xenograft growth. Oncol. Res. 1998, 10, 133–142. [Google Scholar] [PubMed]
- Fung-Leung, W.P.; De Sousa-Hitzler, J.; Ishaque, A.; Zhou, L.; Pang, J.; Ngo, K.; Panakos, J.A.; Chourmouzis, E.; Liu, F.T.; Lau, C.Y. Transgenic mice expressing the human high-affinity immunoglobulin (Ig) E receptor alpha chain respond to human IgE in mast cell degranulation and in allergic reactions. J. Exp. Med. 1996, 183, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef]
- Kayaba, H.; Dombrowicz, D.; Woerly, G.; Papin, J.-P.; Loiseau, S.; Capron, M. Human Eosinophils and Human High Affinity IgE Receptor Transgenic Mouse Eosinophils Express Low Levels of High Affinity IgE Receptor, but Release IL-10 upon Receptor Activation. J. Immunol. 2001, 167, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrowicz, D.; Quatannens, B.; Papin, J.P.; Capron, A.; Capron, M. Expression of a functional Fc epsilon RI on rat eosinophils and macrophages. J. Immunol. 2000, 165, 1266–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Bae, J.S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Duluc, D.; Corvaisier, M.; Blanchard, S.; Catala, L.; Descamps, P.; Gamelin, E.; Ponsoda, S.; Delneste, Y.; Hebbar, M.; Jeannin, P. Interferon-γ reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int. J. Cancer 2009, 125, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Y.; Luo, R.Z.; Peng, R.J.; Wang, S.S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Pellizzari, G.; Bax, H.J.; Josephs, D.H.; Gotovina, J.; Jensen-Jarolim, E.; Spicer, J.F.; Karagiannis, S.N. Harnessing Therapeutic IgE Antibodies to Re-educate Macrophages against Cancer. Trends Mol. Med. 2020, 26, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Salojin, K.V.; Owusu, I.B.; Millerchip, K.A.; Potter, M.; Platt, K.A.; Oravecz, T. Essential Role of MAPK Phosphatase-1 in the Negative Control of Innate Immune Responses. J. Immunol. 2006, 176, 1899. [Google Scholar] [CrossRef] [Green Version]
- Terajima, M.; Inoue, T.; Magari, K.; Yamazaki, H.; Higashi, Y.; Mizuhara, H. Anti-inflammatory effect and selectivity profile of AS1940477, a novel and potent p38 mitogen-activated protein kinase inhibitor. Eur. J. Pharmacol. 2013, 698, 455–462. [Google Scholar] [CrossRef]
- Josephs, D.H.; Nakamura, M.; Bax, H.J.; Dodev, T.S.; Muirhead, G.; Saul, L.; Karagiannis, P.; Ilieva, K.M.; Crescioli, S.; Gazinska, P.; et al. An immunologically relevant rodent model demonstrates safety of therapy using a tumour-specific IgE. Allergy 2018, 73, 2328–2341. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Mazier, D.; Moynet, D.; Thiolat, D.; Malvy, D.; Mossalayi, M.D. IgE mediates killing of intracellular Toxoplasma gondii by human macrophages through CD23-dependent, interleukin-10 sensitive pathway. PLoS ONE 2011, 6, e18289. [Google Scholar] [CrossRef] [Green Version]
- Paiva, C.N.; Figueiredo, R.T.; Kroll-Palhares, K.; Silva, A.A.; Silverio, J.C.; Gibaldi, D.; Pyrrho Ados, S.; Benjamim, C.F.; Lannes-Vieira, J.; Bozza, M.T. CCL2/MCP-1 controls parasite burden, cell infiltration, and mononuclear activation during acute Trypanosoma cruzi infection. J. Leukoc. Biol. 2009, 86, 1239–1246. [Google Scholar] [CrossRef]
- Marone, G.; Gambardella, A.R.; Mattei, F.; Mancini, J.; Schiavoni, G.; Varricchi, G. Basophils in Tumor Microenvironment and Surroundings. Adv. Exp. Med. Biol. 2020, 1224, 21–34. [Google Scholar] [CrossRef]
- Marone, G.; Varricchi, G.; Loffredo, S.; Granata, F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur. J. Pharmacol. 2016, 778, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Crivellato, E.; Travan, L.; Ribatti, D. Mast cells and basophils: A potential link in promoting angiogenesis during allergic inflammation. Int. Arch. Allergy Immunol. 2010, 151, 89–97. [Google Scholar] [CrossRef]
- Maltby, S.; Khazaie, K.; McNagny, K.M. Mast cells in tumor growth: Angiogenesis, tissue remodelling and immune-modulation. Biochim. Biophys. Acta 2009, 1796, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Galli, S.J.; Tsai, M.; Piliponsky, A.M. The development of allergic inflammation. Nature 2008, 454, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Bax, H.J.; Keeble, A.H.; Gould, H.J. Cytokinergic IgE Action in Mast Cell Activation. Front. Immunol. 2012, 3, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, H.J.; Bowen, H.; Beavil, R.L.; Chung, R.; Ward, M.; Davies, A.M.; Dodev, T.S.; McDonnell, J.M.; Beavil, A.J.; Sutton, B.J.; et al. IgE Trimers Drive SPE-7 Cytokinergic Activity. Sci. Rep. 2017, 7, 8164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, H.J.; Bowen, H.; Dodev, T.S.; Sutton, B.J.; Gould, H.J. Mechanism of the antigen-independent cytokinergic SPE-7 IgE activation of human mast cells in vitro. Sci. Rep. 2015, 5, 9538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, H.; Kinet, J.P. Signalling through the high-affinity IgE receptor Fc epsilonRI. Nature 1999, 402, B24–B30. [Google Scholar] [CrossRef]
- Bax, H.J.; Khiabany, A.; Stavraka, C.; Pellizzari, G.; Chan Wah Hak, C.; Robinson, A.; Ilieva, K.M.; Woodman, N.; Naceur-Lombardelli, C.; Gillett, C.; et al. Basophil activation test in cancer patient blood evaluating potential hypersensitivity to an anti-tumor IgE therapeutic candidate. Allergy 2020, 00, 1–5. [Google Scholar] [CrossRef]
- Bettler, B.; Hofstetter, H.; Rao, M.; Yokoyama, W.M.; Kilchherr, F.; Conrad, D.H. Molecular structure and expression of the murine lymphocyte low-affinity receptor for IgE (Fc epsilon RII). Proc. Natl. Acad. Sci. USA 1989, 86, 7566–7570. [Google Scholar] [CrossRef] [Green Version]
- Kinet, J.P. The high-affinity IgE receptor (Fc epsilon RI): From physiology to pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef] [PubMed]
- Maurer, D.; Fiebiger, E.; Reininger, B.; Wolff-Winiski, B.; Jouvin, M.H.; Kilgus, O.; Kinet, J.P.; Stingl, G. Expression of functional high affinity immunoglobulin E receptors (Fc epsilon RI) on monocytes of atopic individuals. J. Exp. Med. 1994, 179, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.P.; Crescioli, S.; Sow, H.S.; Bax, H.J.; Hobbs, C.; Ilieva, K.M.; French, E.; Pellizzari, G.; Cox, V.; Josephs, D.H.; et al. In vivo safety profile of a CSPG4-directed IgE antibody in an immunocompetent rat model. MAbs 2020, 12, 1685349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saul, L.; Josephs, D.H.; Cutler, K.; Bradwell, A.; Karagiannis, P.; Selkirk, C.; Gould, H.J.; Jones, P.; Spicer, J.F.; Karagiannis, S.N. Comparative reactivity of human IgE to cynomolgus monkey and human effector cells and effects on IgE effector cell potency. MAbs 2014, 6, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, M.I.; Silva-Carvalho, R.; Pires, I.; Prada, J.; Bianchini, R.; Jensen-Jarolim, E.; Queiroga, F.L. A Comparative Approach of Tumor-Associated Inflammation in Mammary Cancer between Humans and Dogs. Biomed. Res. Int. 2016, 2016, 4917387. [Google Scholar] [CrossRef]
- Singer, J.; Jensen-Jarolim, E. IgE-based Immunotherapy of Cancer-A Comparative Oncology Approach. J. Carcinog. Mutagen. 2014, 5, 1000176. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, I.; Gotovina, J.; Fazekas-Singer, J.; Fischer, M.B.; Hufnagl, K.; Bianchini, R.; Jensen-Jarolim, E. Canine macrophages can like human macrophages be in vitro activated toward the M2a subtype relevant in allergy. Dev. Comp. Immunol. 2018, 82, 118–127. [Google Scholar] [CrossRef]
- Ilieva, K.M.; Cheung, A.; Mele, S.; Chiaruttini, G.; Crescioli, S.; Griffin, M.; Nakamura, M.; Spicer, J.F.; Tsoka, S.; Lacy, K.E.; et al. Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front. Immunol. 2017, 8, 1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomp, R.; Hensbergen, P.J.; Rombouts, Y.; Zauner, G.; Dragan, I.; Koeleman, C.A.; Deelder, A.M.; Wuhrer, M. Site-specific N-glycosylation analysis of human immunoglobuline. J. Proteome Res. 2014, 13, 536–546. [Google Scholar] [CrossRef]
- Plomp, R.; Bondt, A.; de Haan, N.; Rombouts, Y.; Wuhrer, M. Recent Advances in Clinical Glycoproteomics of Immunoglobulins (Igs). Mol. Cell Proteom. 2016, 15, 2217–2228. [Google Scholar] [CrossRef] [Green Version]
- Shade, K.T.; Conroy, M.E.; Anthony, R.M. IgE Glycosylation in Health and Disease. Curr. Top. Microbiol. Immunol. 2019, 423, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.T.; Platzer, B.; Washburn, N.; Mani, V.; Bartsch, Y.C.; Conroy, M.; Pagan, J.D.; Bosques, C.; Mempel, T.R.; Fiebiger, E.; et al. A single glycan on IgE is indispensable for initiation of anaphylaxis. J. Exp. Med. 2015, 212, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epp, A.; Sullivan, K.C.; Herr, A.B.; Strait, R.T. Immunoglobulin Glycosylation Effects in Allergy and Immunity. Curr. Allergy Asthma Rep. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.C.; Conroy, M.E.; Washburn, N.; Kitaoka, M.; Huynh, D.J.; Laprise, E.; Patil, S.U.; Shreffler, W.G.; Anthony, R.M. Sialylation of immunoglobulin E is a determinant of allergic pathogenicity. Nature 2020, 582, 265–270. [Google Scholar] [CrossRef]
- Blundell, P.A.; Lu, D.; Dell, A.; Haslam, S.; Pleass, R.J. Choice of Host Cell Line Is Essential for the Functional Glycosylation of the Fc Region of Human IgG1 Inhibitors of Influenza B Viruses. J. Immunol. 2020, 204, 1022–1034. [Google Scholar] [CrossRef]
- Chiang, A.W.; Li, S.; Spahn, P.N.; Richelle, A.; Kuo, C.C.; Samoudi, M.; Lewis, N.E. Modulating carbohydrate-protein interactions through glycoengineering of monoclonal antibodies to impact cancer physiology. Curr. Opin. Struct. Biol. 2016, 40, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug. Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Wilson, J.M.; Platts-Mills, T.A.E. IgE to galactose-α-1,3-galactose and the α-Gal syndrome: Insights from basophil activation testing. J. Allergy Clin. Immunol. 2019, 143, 101–103. [Google Scholar] [CrossRef] [Green Version]
- Steinke, J.W.; Platts-Mills, T.A.; Commins, S.P. The alpha-gal story: Lessons learned from connecting the dots. J. Allergy Clin. Immunol. 2015, 135, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Mirakhur, B.; Chan, E.; Le, Q.-T.; Berlin, J.; Morse, M.; Murphy, B.A.; Satinover, S.M.; Hosen, J.; Mauro, D.; et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N. Engl. J. Med. 2008, 358, 1109–1117. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Liu, T.; Yang, L.; Daus, A.; Crowley, R.; Zhou, Q. Structural characterization of N-linked oligosaccharides on monoclonal antibody cetuximab by the combination of orthogonal matrix-assisted laser desorption/ionization hybrid quadrupole-quadrupole time-of-flight tandem mass spectrometry and sequential enzymatic digestion. Anal. Biochem. 2007, 364, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Montero-Morales, L.; Maresch, D.; Castilho, A.; Turupcu, A.; Ilieva, K.M.; Crescioli, S.; Karagiannis, S.N.; Lupinek, C.; Oostenbrink, C.; Altmann, F.; et al. Recombinant plant-derived human IgE glycoproteomics. J. Proteom. 2017, 161, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardor, M.; Faveeuw, C.; Fitchette, A.C.; Gilbert, D.; Galas, L.; Trottein, F.; Faye, L.; Lerouge, P. Immunoreactivity in mammals of two typical plant glyco-epitopes, core alpha(1,3)-fucose and core xylose. Glycobiology 2003, 13, 427–434. [Google Scholar] [CrossRef]
- Ward, B.J.; Landry, N.; Trépanier, S.; Mercier, G.; Dargis, M.; Couture, M.; D’Aoust, M.A.; Vézina, L.P. Human antibody response to N-glycans present on plant-made influenza virus-like particle (VLP) vaccines. Vaccine 2014, 32, 6098–6106. [Google Scholar] [CrossRef] [PubMed]
- Shaaltiel, Y.; Tekoah, Y. Plant specific N-glycans do not have proven adverse effects in humans. Nat. Biotechnol. 2016, 34, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I. Chapter 11-Treatment of Ebola Virus Disease: Therapeutic Agents. In Ebola Virus Disease; Qureshi, A.I., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 159–166. [Google Scholar] [CrossRef]
- Chinuki, Y.; Morita, E. Alpha-Gal-containing biologics and anaphylaxis. Allergol. Int. 2019, 68, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Heinzerling, L.; Mari, A.; Bergmann, K.C.; Bresciani, M.; Burbach, G.; Darsow, U.; Durham, S.; Fokkens, W.; Gjomarkaj, M.; Haahtela, T.; et al. The skin prick test-European standards. Clin. Transl. Allergy 2013, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Wise, S.K.; Lin, S.Y.; Toskala, E.; Orlandi, R.R.; Akdis, C.A.; Alt, J.A.; Azar, A.; Baroody, F.M.; Bachert, C.; Canonica, G.W.; et al. International Consensus Statement on Allergy and Rhinology: Allergic Rhinitis. Int. Forum. Allergy Rhinol. 2018, 8, 108–352. [Google Scholar] [CrossRef]
- Crobach, M.J.; Hermans, J.; Kaptein, A.A.; Ridderikhoff, J.; Petri, H.; Mulder, J.D. The diagnosis of allergic rhinitis: How to combine the medical history with the results of radioallergosorbent tests and skin prick tests. Scand. J. Prim. Health Care 1998, 16, 30–36. [Google Scholar] [CrossRef]
- Verstege, A.; Mehl, A.; Rolinck-Werninghaus, C.; Staden, U.; Nocon, M.; Beyer, K.; Niggemann, B. The predictive value of the skin prick test weal size for the outcome of oral food challenges. Clin. Exp. Allergy 2005, 35, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Leguy-Seguin, V.; Jolimoy, G.; Coudert, B.; Pernot, C.; Dalac, S.; Vabres, P.; Collet, E. Diagnostic and predictive value of skin testing in platinum salt hypersensitivity. J. Allergy Clin. Immunol. 2007, 119, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, K.M.; Rybicki, L.A.; Kennedy, A.W.; Belinson, J.L.; Webster, K.D.; Kulp, B.; Peterson, G.; Markman, M. Carboplatin skin testing: A skin-testing protocol for predicting hypersensitivity to carboplatin chemotherapy. J. Clin. Oncol. 2001, 19, 3126–3129. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Zanotti, K.; Peterson, G.; Kulp, B.; Webster, K.; Belinson, J. Expanded experience with an intradermal skin test to predict for the presence or absence of carboplatin hypersensitivity. J. Clin. Oncol. 2003, 21, 4611–4614. [Google Scholar] [CrossRef] [PubMed]
- Caiado, J.; Venemalm, L.; Pereira-Santos, M.C.; Costa, L.; Barbosa, M.P.; Castells, M. Carboplatin-, oxaliplatin-, and cisplatin-specific IgE: Cross-reactivity and value in the diagnosis of carboplatin and oxaliplatin allergy. J. Allergy Clin. Immunol. Pract. 2013, 1, 494–500. [Google Scholar] [CrossRef]
- Haahtela, T.; Burbach, G.J.; Bachert, C.; Bindslev-Jensen, C.; Bonini, S.; Bousquet, J.; Bousquet-Rouanet, L.; Bousquet, P.J.; Bresciani, M.; Bruno, A.; et al. Clinical relevance is associated with allergen-specific wheal size in skin prick testing. Clin. Exp. Allergy 2014, 44, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Douiri, A.; Becares, N.; Wu, S.Y.; Stephens, A.; Radulovic, S.; Chan, S.M.; Fox, A.T.; Du Toit, G.; Turcanu, V.; et al. Basophil activation test discriminates between allergy and tolerance in peanut-sensitized children. J. Allergy Clin. Immunol. 2014, 134, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.F.; Shreffler, W.G. Road map for the clinical application of the basophil activation test in food allergy. Clin. Exp. Allergy 2017, 47, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Ford, L.S.; Bloom, K.A.; Nowak-Wegrzyn, A.H.; Shreffler, W.G.; Masilamani, M.; Sampson, H.A. Basophil reactivity, wheal size, and immunoglobulin levels distinguish degrees of cow’s milk tolerance. J. Allergy Clin. Immunol. 2013, 131, 180–186. [Google Scholar] [CrossRef] [Green Version]
- Ocmant, A.; Mulier, S.; Hanssens, L.; Goldman, M.; Casimir, G.; Mascart, F.; Schandene, L. Basophil activation tests for the diagnosis of food allergy in children. Clin. Exp. Allergy 2009, 39, 1234–1245. [Google Scholar] [CrossRef]
- Salas, M.; Fernandez-Santamaria, R.; Mayorga, C.; Barrionuevo, E.; Ariza, A.; Posadas, T.; Laguna, J.J.; Montanez, M.I.; Molina, N.; Fernandez, T.D.; et al. Use of the Basophil Activation Test May Reduce the Need for Drug Provocation in Amoxicillin-Clavulanic Allergy. J. Allergy Clin. Immunol. Pract. 2018, 6, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.J.; Padial, A.; Mayorga, C.; Fernandez, T.; Sanchez-Sabate, E.; Cornejo-Garcia, J.A.; Antunez, C.; Blanca, M. The diagnostic interpretation of basophil activation test in immediate allergic reactions to betalactams. Clin. Exp. Allergy 2004, 34, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Mayorga, C.; Ariza, A.; Dona, I.; Rosado, A.; Blanca-Lopez, N.; Andreu, I.; Torres, M.J. In vitro evaluation of IgE-mediated hypersensitivity reactions to quinolones. Allergy 2011, 66, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Laguna, J.J.; Bogas, G.; Salas, M.; Mayorga, C.; Dionicio, J.; Gonzalez-Mendiola, R.; Ariza, A.; Fernandez-Santamaria, R.; Olazabal, I.; Dona, I.; et al. The Basophil Activation Test Can Be of Value for Diagnosing Immediate Allergic Reactions to Omeprazole. J. Allergy Clin. Immunol. Pract. 2018, 6, 1628–1636. [Google Scholar] [CrossRef]
- Erdmann, S.M.; Sachs, B.; Kwiecien, R.; Moll-Slodowy, S.; Sauer, I.; Merk, H.F. The basophil activation test in wasp venom allergy: Sensitivity, specificity and monitoring specific immunotherapy. Allergy 2004, 59, 1102–1109. [Google Scholar] [CrossRef]
- Sturm, G.J.; Bohm, E.; Trummer, M.; Weiglhofer, I.; Heinemann, A.; Aberer, W. The CD63 basophil activation test in Hymenoptera venom allergy: A prospective study. Allergy 2004, 59, 1110–1117. [Google Scholar] [CrossRef]
- Bokanovic, D.; Arzt-Gradwohl, L.; Schwarz, I.; Schrautzer, C.; Laipold, K.; Aberer, W.; Binder, B.; Sturm, G.J. Possible utility of basophil activation test in dual honeybee and vespid sensitization. J. Allergy Clin. Immunol. Pract. 2020, 8, 392–394. [Google Scholar] [CrossRef]
- Hemmings, O.; Kwok, M.; McKendry, R.; Santos, A.F. Basophil Activation Test: Old and New Applications in Allergy. Curr. Allergy Asthma Rep. 2018, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Giavina-Bianchi, P.; Galvao, V.R.; Picard, M.; Caiado, J.; Castells, M.C. Basophil Activation Test is a Relevant Biomarker of the Outcome of Rapid Desensitization in Platinum Compounds-Allergy. J. Allergy Clin. Immunol. Pract. 2017, 5, 728–736. [Google Scholar] [CrossRef]
- Iwamoto, T.; Hirai, H.; Yamaguchi, N.; Kobayashi, N.; Sugimoto, H.; Tabata, T.; Okuda, M. Carboplatin-induced severe hypersensitivity reaction: Role of IgE-dependent basophil activation and FcepsilonRI. Cancer Sci. 2014, 105, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Sugimoto, H.; Tabata, T.; Okuda, M. Clinical Utility of Basophil CD203c as a Biomarker for Predicting the Timing of Hypersensitivity Reaction in Carboplatin Rechallenge: Three Case Reports. Clin. Ther. 2016, 38, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Yuta, A.; Tabata, T.; Sugimoto, H.; Gabazza, E.C.; Hirai, H.; Kojima, S.; Okuda, M. Evaluation of basophil CD203c as a predictor of carboplatin-related hypersensitivity reaction in patients with gynecologic cancer. Biol. Pharm. Bull. 2012, 35, 1487–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornelas, C.; Caiado, J.; Campos Melo, A.; Pereira Barbosa, M.; Castells, M.C.; Pereira Dos Santos, M.C. The Contribution of the Basophil Activation Test to the Diagnosis of Hypersensitivity Reactions to Oxaliplatin. Int. Arch. Allergy Immunol. 2018, 177, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.F.; Basu, B.; Montes, A.; Banerji, U.; Kristeleit, R.; Veal, G.J.; Corrigan, C.; Till, S.J.; Nintos, G.; Brier, T.; et al. Phase 1 trial of MOv18, a first-in-class IgE antibody therapy for cancer. In Proceedings of the AACR Annual Meeting 2020, VPO.CT01, Los Angeles, CA, USA, 24–29 April 2020. [Google Scholar]
- Barton, C.; Vigor, K.; Scott, R.; Jones, P.; Lentfer, H.; Bax, H.J.; Josephs, D.H.; Karagiannis, S.N.; Spicer, J.F. Beta-glucan contamination of pharmaceutical products: How much should we accept? Cancer Immunol. Immunother. 2016, 65, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigor, K.; Emerson, J.; Scott, R.; Cheek, J.; Barton, C.; Bax, H.J.; Josephs, D.H.; Karagiannis, S.N.; Spicer, J.F.; Lentfer, H. Development of downstream processing to minimize beta-glucan impurities in GMP-manufactured therapeutic antibodies. Biotechnol. Prog. 2016, 32, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target of IgE | IgE Species | Direct Effect | ADCC | ADCP | Degranulation 1 | Antigen Presentation | Refs. |
|---|---|---|---|---|---|---|---|
| Human FRα | Mouse/human chimeric | - | ✓ | ✓ | ✓ | - | [29,30,42,49] |
| Human FRα | Mouse/rat chimeric | - | ✓ | ✓ | - | - | [32] |
| Human Her2 | Humanised | ✓ | ✓ | ✕ | ✓ | - | [36,43] |
| Human EGFR | Mouse/human chimeric | ✓ | ✓ | ✕ | ✓ | - | [44] |
| Human Her2 | Human | - | - | - | ✓ | ✓ | [47] |
| Human PSA | Mouse/human chimeric | - | - | - | ✓ | ✓ | [48] |
| Human CD20 | Mouse/human chimeric | - | ✓ | ✕ | - | - | [45] |
| Human colorectal cancer | Human | - | ✓ | ✕ | - | - | [7] |
| Human EGFR | Canine | - | ✓ | ✕ | - | - | [37] |
| Human Her2 | Human 2 | - | - | - | ✓ | - | [39] |
| Human SF-25 | Mouse/human chimeric | - | ✓ | ✕ | - | - | [46] |
| Target of IgE | IgE Species | Tumour Model (Tumour Site) | Animal Model 2 | Antibody Injection Route | Findings 1 | Ref. |
|---|---|---|---|---|---|---|
| Gp36 of MMTV | Mouse | H2712 mouse mammary carcinoma (s.c. and i.p.) | Syngeneic C3H/HeJ mice | i.p. | Inhibited the development of tumours | [50] |
| COLO 205 | Mouse and mouse/human chimeric | Human colorectal COLO 205 carcinoma (s.c.) | SCID mice | i.v | Inhibited the growth of established tumours | [51] |
| Human FRα | Mouse/human chimeric | IGROV1 human ovarian carcinoma (s.c.) | PBMC engrafted SCID mice | i.v. | Greater anti-tumour activity than IgG1 | [31] |
| HUA patient derived ovarian carcinoma donor mice ascites (i.p.) | PBMC engrafted nude mice | i.p. | Prolonged mouse survival 1 | [29] | ||
| PBMC 2 engrafted nude mice | i.p. | Prolonged mouse survival 1 | [42] | |||
| U937 (+/− IL-4 treatment) engrafted nude mice | i.p. | Prolonged mouse survival 1 | [30] | |||
| Human Her2 | Human | Her2-expressing D2F2/E2 mouse mammary carcinoma (i.p.) | hFceR1α Tg BALB/c mice | i.p. | Prolonged mouse survival | [47] |
| Human MUC1 | Mouse/human chimeric | MUC1-expressing 4T1 mouse mammary carcinoma (s.c.) | hFceR1α Tg BALB/c mice | s.c | Reduced tumour growth by 25–30% | [45] |
| Human PSA | Mouse/human chimeric | PSA-expressing CT26 mouse colon adenocarcinoma (s.c.) | hFceR1α Tg BALB/c mice | s.c | Prolonged mouse survival | [48] |
| Human FRα | Mouse/rat chimeric | FRα-expressing CC531 rat colon adenocarcinoma (lung mets. from i.v. injection) | Immuno-competent WAG rat | i.v. | Superior efficacy compared to mouse/rat IgG2b | [32] |
| Therapeutic Antibody | Antibody Target | Dose | Model Species | Findings | Ref. |
|---|---|---|---|---|---|
| C6MH3-B1 IgE | Human Her2 | 2.4 and 80 µg/kg | Cynomolgus monkey | No systemic reactions or adverse events observed. | [47] |
| MOv18 IgE | Human FRα | 5, 10, 50 mg/kg | Rat | No clinical, histopathological or metabolic signs of a type I hypersensitivity reaction, but some mild responses such as piloerection and hunching which were observed to the same degree in IgG-treated rats. | [67] |
| CSPG4 IgE | Human and rat CSPG4 | 10 mg/kg (for immediate reactions); 5 mg/kg (for long-term safety) | Rat | Only transient mild to moderate adverse events within 5 min which resolved by 30 min, accompanied by mild elevation in serum tryptase. No long-term toxicity observed. | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chauhan, J.; McCraw, A.J.; Nakamura, M.; Osborn, G.; Sow, H.S.; Cox, V.F.; Stavraka, C.; Josephs, D.H.; Spicer, J.F.; Karagiannis, S.N.; et al. IgE Antibodies against Cancer: Efficacy and Safety. Antibodies 2020, 9, 55. https://doi.org/10.3390/antib9040055
Chauhan J, McCraw AJ, Nakamura M, Osborn G, Sow HS, Cox VF, Stavraka C, Josephs DH, Spicer JF, Karagiannis SN, et al. IgE Antibodies against Cancer: Efficacy and Safety. Antibodies. 2020; 9(4):55. https://doi.org/10.3390/antib9040055
Chicago/Turabian StyleChauhan, Jitesh, Alex J. McCraw, Mano Nakamura, Gabriel Osborn, Heng Sheng Sow, Vivienne F. Cox, Chara Stavraka, Debra H. Josephs, James F. Spicer, Sophia N. Karagiannis, and et al. 2020. "IgE Antibodies against Cancer: Efficacy and Safety" Antibodies 9, no. 4: 55. https://doi.org/10.3390/antib9040055