1. Introduction
The advanced oxidation processes (AOPs) are used in drinking water and both municipal and industrial wastewater purification to transform organic pollutants into less toxic by-products [
1]. Among many AOPs, photoelectrocatalysis is an emerging and promising hybrid AOP [
2]. This technology provides synergistic benefits from the interaction between photocatalytic and electrocatalytic processes [
3,
4]. Similar to photocatalysis, photoelectrocatalysis relies on semiconductor materials to photo-generate charge carriers according to Equation (1); applying a constant voltage across semiconductors supported on solid surfaces prevents recombination within the material [
5,
6]. The photoexcitation of electrons from the filled semiconductor valence band to the empty conduction band generates charge carriers when irradiated with photons of energy superior to the band gap (E
g) [
7,
8]. Electron (e
cb−) photoexcitation also generates a vacancy at the valence band (h
vb+). The h
vb+ are highly–oxidizing species that can mineralize organic compounds or yield reactive oxygen species (ROS), such as hydroxyl radicals (
•OH), from water oxidation according to Equation (2) [
9,
10,
11].
Charge carriers are unstable due to their excited state and tend to recombine according to Equation (3), which may drastically diminish the availability of oxidants on the semiconductor surface [
12,
13]. The main contribution of the electrochemically-driven component in the photoelectrocatalytic process is to diminish the extent and even avoid recombination [
14,
15]. This is achieved by imposing a difference of potential during photoexcitation to induce separation of photogenerated charge carriers [
2,
16].
Pollutant degradation in water dramatically increases when semiconductors are employed as photoanodes [
2,
17,
18]. Unlike in slurry reactors, photocatalysts must be mounted on electrically-conducting surfaces. Therefore, a major barrier for photoelectrocatalytic reactors is the surface area of electrodes that can be coated with photocatalysts [
19,
20]. Previous approaches have considered irradiation outside the cell through transparent conductive glass electrodes of indium tin oxide and fluorine-doped tin oxide (ITO/FTO) coated with semiconductor photocatalysts. However, this approach faces (i) electrode stability problems associated with leaching of the coatings and (ii) photon transport losses due to the absorption by glass [
21,
22,
23]. Herein, a promising alternative reactor design is presented that uses a desirable parallel electrode arrangement in a single annular reactor compartment to promote a homogeneous current distribution [
20,
24]. Illustrated in
Figure 1, multiple disc-shaped photoanodes and cathodes were aligned perpendicular to an annular ultraviolet (UV) lamp placed in the middle of the reactor. This novel reactor design aims to reconcile the needs of electrochemical systems that ensure homogeneous distribution of current density by using parallel electrodes and enabling photoexcitation through efficient light delivery. Removal and mineralization of acetaminophen, a commonly occurring pharmaceutical in treated municipal wastewater [
25,
26,
27], was evaluated in the reactor. This reactor uses electrical energy per order (E
EO, kWh m
−3 order
−1), to assess performance. E
EO is the energy required for 90% pollutant degradation and includes energy for both the photon-driven and electrically-driven processes used in this reactor. Oxidation by-products were identified by high-performance liquid chromatography (HPLC), total organic carbon, and total nitrogen analyses. Comparing photocatalytic to photoelectrocatalytic performance at different applied currents and initial acetaminophen concentrations showed that performance depended on light intensity reaching the electrode surfaces.
2. Materials and Methods
2.1. Electrochemical Cell and Photoelectrode Synthesis
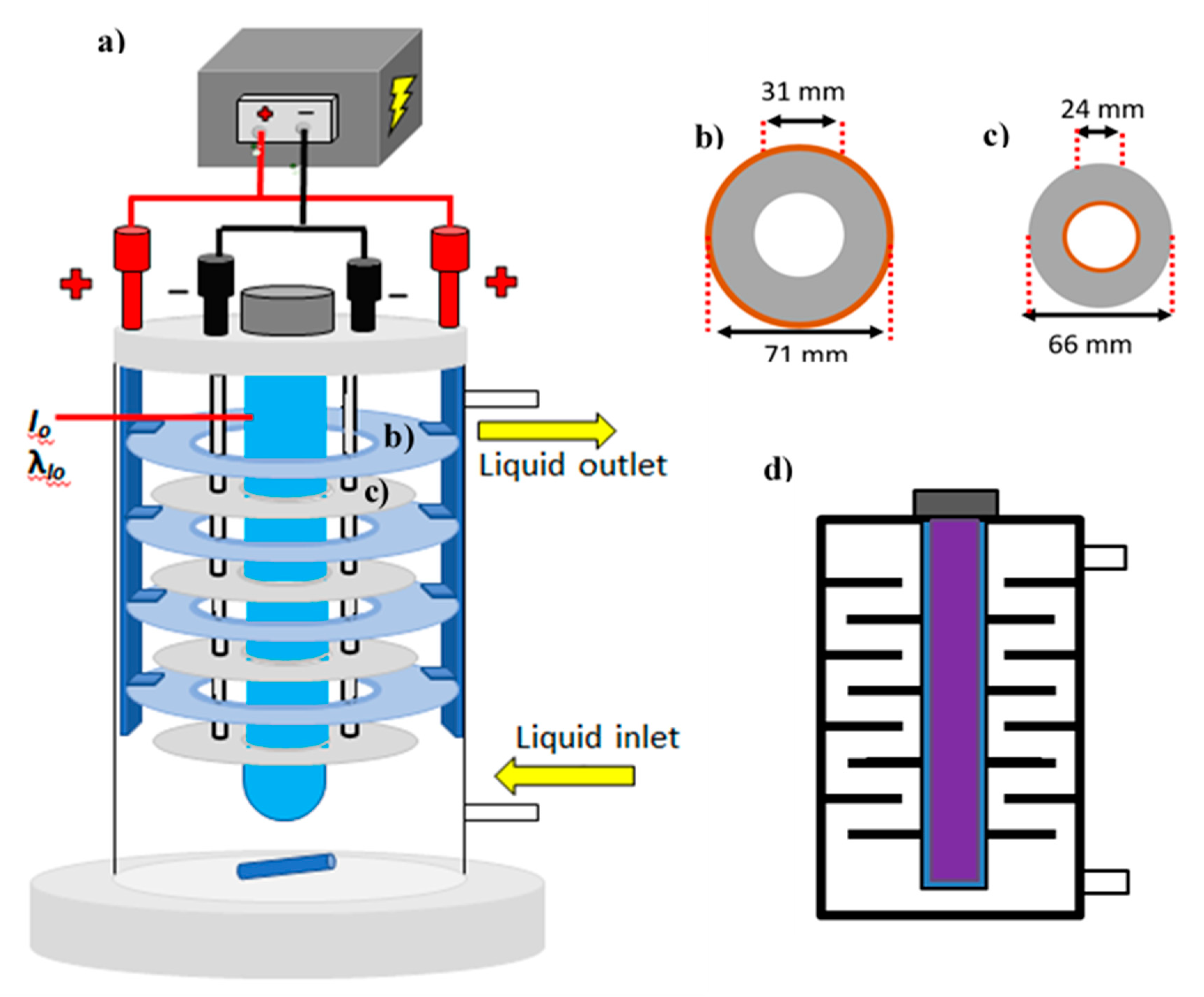
Figure 1 shows the custom-made photoelectrocatalytic reactor configuration used to treat 1 L solutions containing 5 to 50 mg L
−1 of acetaminophen. Eight donut-shaped electrode discs were aligned perpendicular to a 14 W UV lamp GPH287T5L/4 (λ = 275 nm; emission spectra shown in
Figure 2) encased in a quartz tube (YUP, China). The electrodes were operated potentiostatically using a Tenma 72-8340A potentiostat/galvanostat (Tenma, US). A Masterflex L/S peristaltic pump (Cole-Parmer, US) circulated the solution at 180 mL min
−1 to provide mixing and to overcome mass transport limitations of acetaminophen to the reactive surfaces on the donut-shaped disc electrodes.
As depicted in
Figure 1b, the donut-shaped discs used as photo-anodes had a total diameter of 71 mm with a defined surface area of 32 cm
2 per side. The position of the electrodes ensured homogeneous current distribution while ensuring light delivery. Note that the architecture of the system defines hydraulic channels for homogeneous distribution of the solution, whereas the incidence of photons emitted from the immersed light source induces photoexcitation to generate charge carriers. The titanium discs were anodized to produce TiO
2 nanotubes on the surface using previously described methods [
28]. Four separate, non-treated, 66 mm in diameter titanium discs were used as cathodes (30 cm
2 surface area per side). Monopolar connection was attained by using titanium foil (99.99%) as a connector bridge between plates of identical polarity. As shown in the sectional view of the cell (
Figure 1d), the electrode configuration allowed a hydraulic pathway during recirculation that maximized solution contact with the electrodes.
Kinetic analysis of acetaminophen degradation followed pseudo-first-order kinetics to determine rate constants (
k1, s
−1). Experiments were conducted in triplicate and showed excellent reproducibility. As shown in Equation (4), the rate constant was used to calculate E
EO [
29], which is a figure of merit defined by the International Union of Pure and Applied Chemistry (IUPAC) to benchmark AOPs as water treatment technologies in terms of their energy requirements to diminish a pollutant’s content by one order of magnitude (i.e., 90% pollutant removal):
where the 6.39 × 10
−4 constant accounts for conversion factor (1 h/3600 s/0.4343); Pl
amp and P
cell are the rated power of the lamp and the electrochemical cell (W), respectively; V
S is the solution volume (L); and
k1 is the rate constant (s
−1).
Photoanode preparation involved polishing Ti discs with sandpaper of grain sizes P600, P1200, and P2000. Discs were degreased under ultrasonication with three polar solvents for 10 min each: (i) methanol, (ii) isopropyl alcohol, and (iii) acetone. Nanotubes were grown on both sides of the electrode by conducting anodization with two Ti disc cathodes of identical dimensions placed at each side of the anode with 1 cm of interelectrode gap. Anodization was conducted potentiostatically by applying 40 V for 2 h in a solution of ethylene glycol containing 0.3% NH
4F and 2 vol. % of water [
28]. The grown nanotubes were thermally annealed at 450 °C for 2 h with a heating ramp of 10 °C min
−1 in the muffle furnace Tuve Furnace 21100 (SentroTech, US). Thermal treatment at 450 °C leads to anatase structure formation [
30]. Synthesized photoanodes were then implemented in the photoelectrocatalytic cell.
2.2. Chemicals
Acetaminophen (99% pure neat powder) and other carboxylic acid standards were purchased from Sigma-Aldrich.
Figure 2 shows the UV–vis spectrum of acetaminophen. Sodium nitrate and ammonium chloride were used as ionic chromatography standards, and sodium sulfate was used as a supporting electrolyte. Analytical grade sulfuric acid and/or sodium hydroxide from Fisher Chemical were employed to adjust the pH. HPLC-grade acetonitrile was purchased from Sigma-Aldrich. Solutions and mobile phases were prepared with nano-pure water obtained from a Millipore Milli-Q system with resistivity >18.2 MΩ cm at 25 °C.
2.3. Analytical Procedures
Scanning electron microscopy (SEM) images of the synthesized TiO2 nanotubes were recorded using a Jeol JSM-6510LV series (JEOL, US). The Raman spectra was recorded using a SENTERRA II Compact Raman Microscope (Bruker, US) with a laser radiation at 532 nm. Photon irradiance, expressed in μW cm−2, was measured with a radiometer Avantes AvaSpec 2048 (Avantes, US) spectrometer with a cosine corrector. Linear sweep voltammetry analyses with 100 mV s−1 scan rates were conducted in a three-electrode, one-compartment cell using a PGSTAT302n potentiostat-galvanostat from Metrohm controlled by Autolab Nova 2.1 software (Metrohm, US). The TiO2 nanotube anode was the working electrode, Pt wire was the counter electrode, and an Ag/AgCl was used as reference electrode.
The pH of the solutions was measured using a Thermo Scientific Orion Star A221 pH meter (Thermo, US). Aliquots were withdrawn during experiments and analyzed. Chromatographic analysis of 25 μL of sample allowed identification and quantification of acetaminophen and by-products (e.g., carboxylic acids, nitrate, and ammonium). Acetaminophen decay was followed by reversed-phase chromatography using a Waters 2695 HPLC coupled to a Waters 2996 Photodiode Array detector, fitted with a Waters LiChrosorb® 10 μm RP18 (100 mm × 4.6 mm) column at 25 °C and a LiChroCART® 4-4 guard column (Waters, US). Chromatograph was operated at isocratic flow using a 35:65 (v/v) acetonitrile/water mixture at 0.6 ml min−1 as mobile phase.
Carboxylic acids yielded during acetaminophen aromatic moiety breakage were identified and quantified by ion-exclusion chromatography using the instrument described above fitted with a Bio-Rad Amnex HPX87H (300 × 7.8 mm) column at 35 °C with a 4.0 mM H2SO4 mobile phase at 0.6 mL min−1(Bio-Rad, US). Nitrogenized organic ion formation was quantified by ionic chromatography using a Thermo Dionex ICS-5000DC coupled to a conductivity detector AERS 500 (Thermo, US). Anionic species nitrite and nitrate were quantified using a high capacity hydroxide-selective anion-exchange column Dionex Ionpac AS18 (2 × 250 mm) flowing 30 mM KOH solution as the mobile phase at 0.25 mL min−1 (Thermo, US). Ammonium was quantified using an ammonia TNT plus Vial Test from Hach with 0.015 to 2.00 mg L−1 NH3-N quantification range.
Mineralization of organics load in treated solutions was evaluated through dissolved organic carbon (DOC) abatement determined with a Shimadzu VCSN total organic carbon (TOC) analyzer. Total nitrogen was measured with a Shimadzu TNM-1 module coupled to the above TOC analyzer.
3. Results and Discussion
3.1. TiO2 Nanotube Photoelectrode Discs’ Characterization
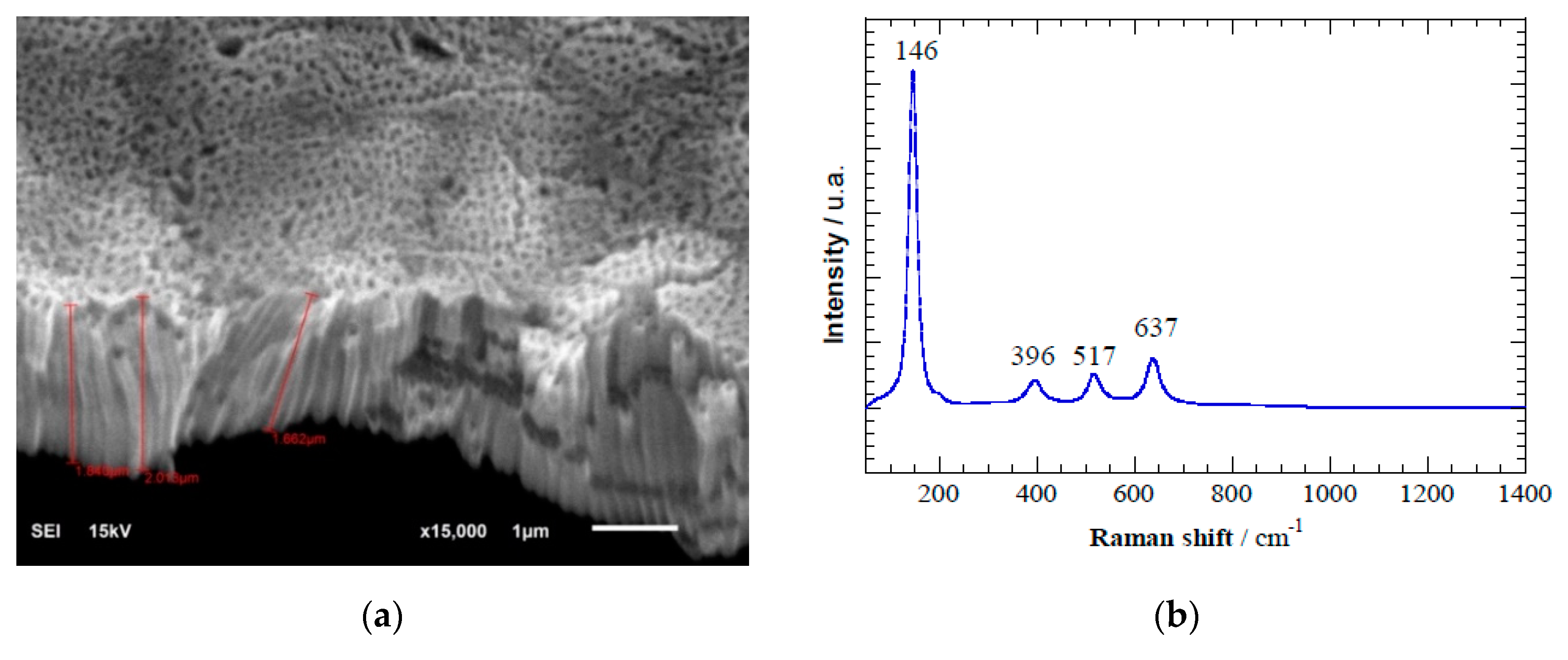
Figure 3a shows the SEM image of hierarchically organized TiO
2 nanotubes after anodization of pure Ti discs. Homogeneous and well-distributed TiO
2 nanotubes of length 1.8–2.0 μm and diameter ≈ 80 nm were perpendicular to the Ti surface. Raman spectroscopy (
Figure 3b) revealed peaks at 146 cm
−1, 396 cm
−1, 517 cm
−1, and 637 cm
−1, which is characteristic of anatase nanotubes [
31]. Anatase formation by the thermal treatment at 450 °C is consistent with literature [
30,
32]. The diffractogram of the anodized TiO
2 (see
Figure 3c) also depicts the characteristic diffraction peaks associated to anatase crystalline phase, which is in agreement with the conclusion obtained from the Raman spectroscopic analysis. The band gap of 3.2 eV determined is coincident with those previously reported in literature for anatase [
33,
34].
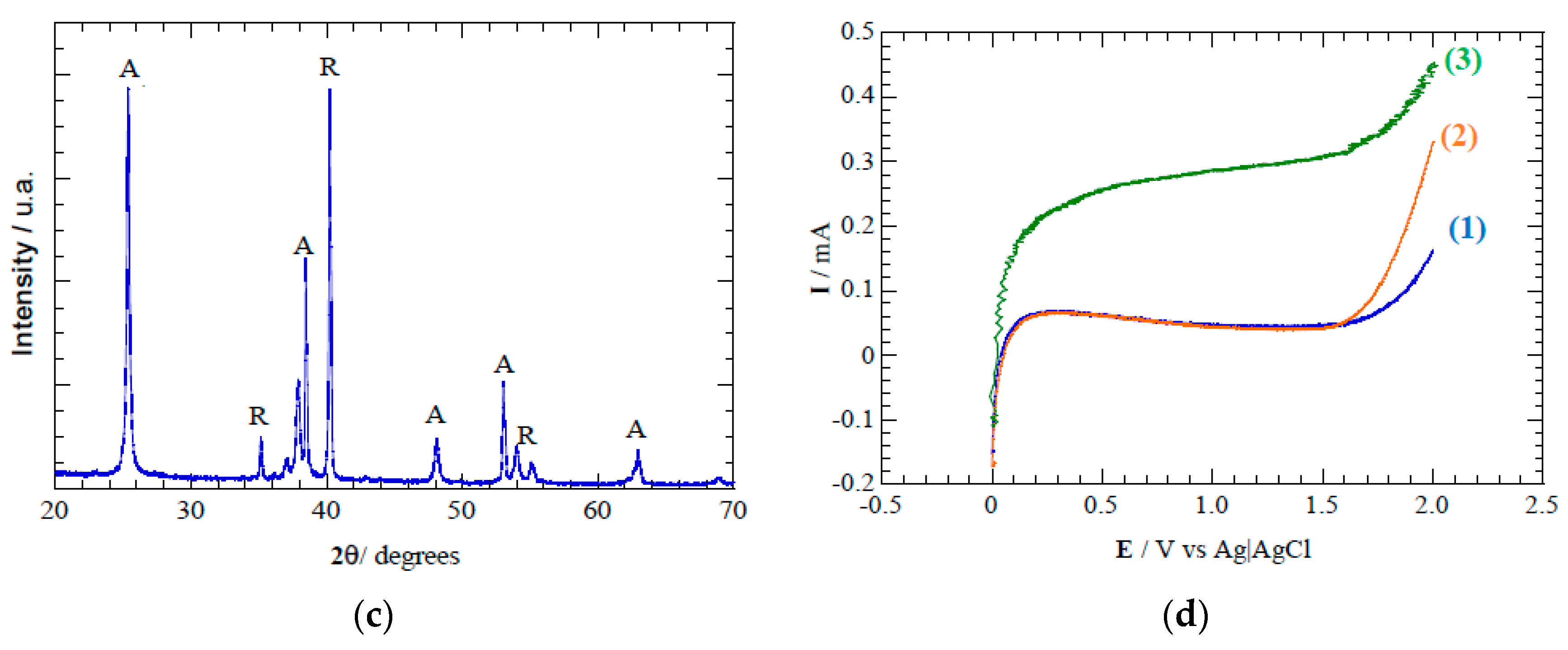
Voltammetric analysis shows an increased current response of the TiO
2 nanotube discs when the photoanode is irradiated (
Figure 3), which is associated with photogeneration of charge carriers [
35,
36,
37]. Photons delivered from the UV light source to the TiO
2 photoanode surface provide the energy required to overcome the band gap (Eg = 3.2 eV) [
38,
39]. The small overpotential of oxygen evolution with an onset potential of 1.65 V vs Ag/AgCl suggests a low capability for
•OH electrogeneration from water oxidation (Equation (5)). Therefore, the acetaminophen does not undergo oxidation by direct charge transfer processes within the electrochemical window of TiO
2 anodes. Indeed, the higher TiO
2 oxidation states stabilize ROS as chemisorbed reactive oxides that may induce electrochemical transformation of acetaminophen, but further mineralization is unlikely due to lower oxidation power of chemisorbed
•OH [
40]. The behavior we observed is characteristic of active anode materials according to Comninellis classification [
41]. These results prove the photoelectrocatalytic activity of the anodes employed in the multi-electrode multi-disc reactor under a monopolar connection (see
Figure 1).
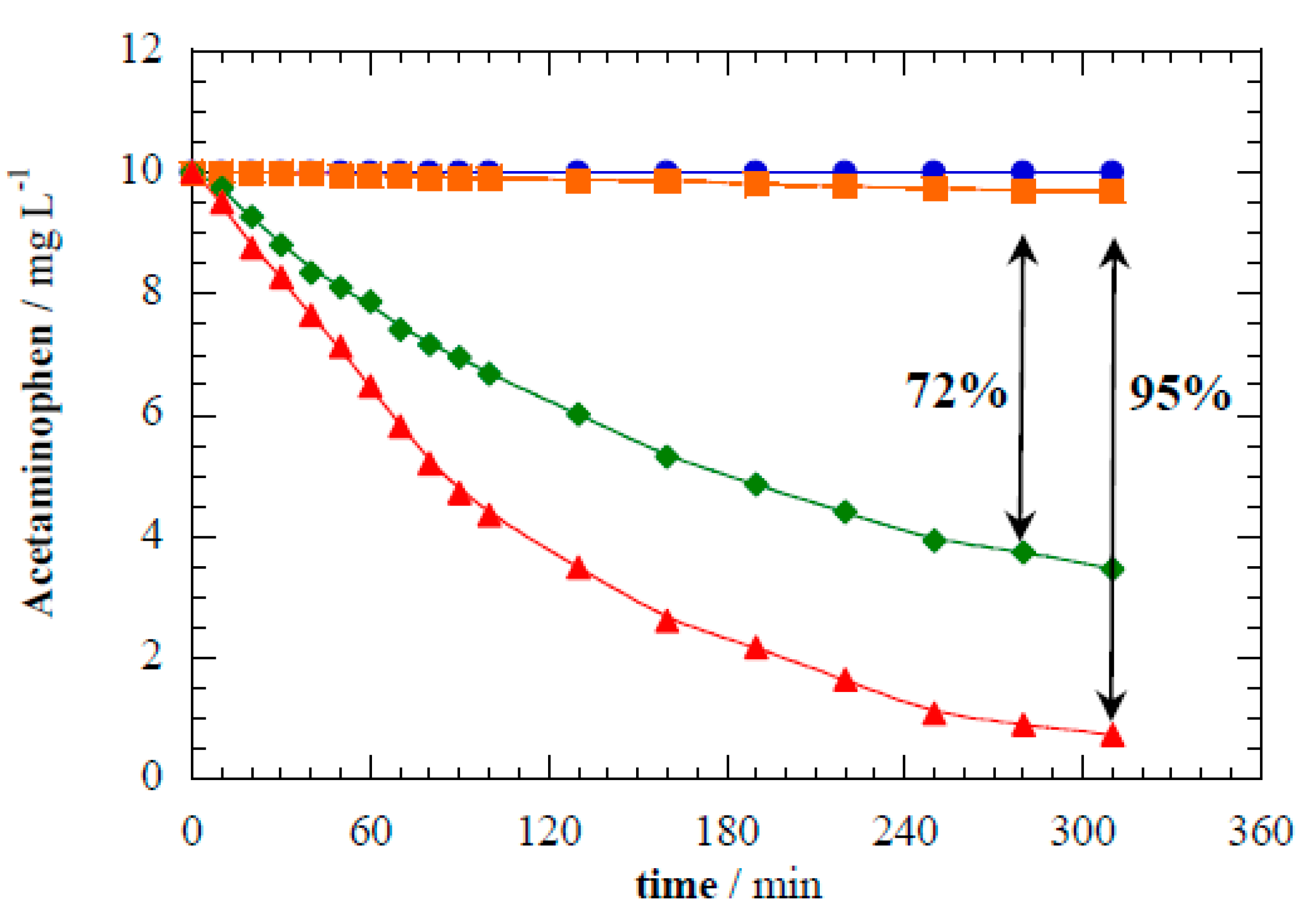
3.2. Acetaminophen Degradation by Photocatalysis, Electrocatalysis, and Photoelectrocatalysis
Figure 4 illustrates the acetaminophen loss kinetics under different reactor operational modes. Direct photolysis of acetaminophen (i.e., without disc electrodes) resulted in negligible degradation after 5 h of UV irradiation. Acetaminophen has a very low quantum yield (Φ
253 = 0.006) and is known to be photostable [
42]. Electrocatalytic oxidation of acetaminophen in the dark under constant cell potential of 8.0 V attained only 3% removal by heterogeneous reactions.
Figure 4 shows that photocatalysis with TiO
2 nanotubes abated 72% acetaminophen after 5 h. This proves that perpendicular alignment of the discs to the lamp supports photocatalysis. The acetaminophen photocatalytic abatement was well fit by first-order kinetics, and yielded a rate constant (
k1) of 6.88 × 10
−5 s
−1 (
R2 = 0.998). As well demonstrated in literature, acetaminophen degradation in the TiO
2 photoelectrocatalytic system is mediated by the oxidation capability of photogenerated h
vb+ and
•OH by reactions seen in Equations (1) and (2) [
7,
8], respectively.
The rate constant for acetaminophen removal in photoelectrocatalytic operational mode (
k1 of 1.42 × 10
−4 s
−1) was nearly 3× higher than in photocatalytic mode. Greater than 95% acetaminophen abatement was attained after 5 h under photoelectrocatalytic treatment at E
cell = 8.0 V (
Figure 4). The photoelectrocatalysis
k1 was over seven orders of magnitude greater than for electrocatalytic treatment alone. Applying a bias potential enhances the charge carrier separation and diminishes the extent of recombination (Equation (3)) [
2,
43], which increases the half-life of the oxidants photogenerated on the photoanode surface [
13,
44]. Moreover, the evolution of O
2 on the anode surface may contribute to selective scavenging e
cb− through Equation (6), which yields superoxide radical (O
2•−) [
16,
45]. Yielded O
2•− is a weaker oxidant with E° = 0.94 V versus a standard hydrogen electrode (SHE) [
46], which may contribute to the acetaminophen degradation by (i) selective oxidation of organics and (ii) enhances charge carriers’ stability. Note that the consumption of e
cb− by Equation (7) avoids the risk of recombination Equation (3). The interaction of photocatalytically and electrochemically-driven processes considerably enhances the photoelectrocatalytic treatment performance.
The higher efficiency of the photoelectrocatalytic system is not only reflected from rate constants but also on energy requirements. Accounting for both lamp and cell power requirements (Equation (4)), EEO requirement for photocatalysis alone is 130 kWh m−3 order−1 compared with 67 kWh m−3 order−1 for photoelectrocatalytic treatment, indicating a reduction by two times in energy requirement. Thus, applying a small bias potential is a promising alternative to increase performance of supported photocatalysts.
3.3. Influence of Applied Cell Potential on Reactor Performance.
As supported by above findings, applying a difference of potential synergistically enhanced performance. The h
vb+ lifetime on the photoanode surface increases because recombination is inhibited (equation (3)) [
19,
39]. Applying a constant cell potential (E
cell) promotes extraction of photo-excited e
cb− through an external electrical circuit in an electrochemically-driven process [
2]. The E
cell defines the efficient transport and consequent charge carrier separation in the photoelectrocatalytic system [
16,
47]. Thus, impacts on photoelectrocatalytic
k1 were studied by varying applied E
cell while maintaining a constant photon flux.
Figure 5 shows increasing degradation kinetics with higher E
cell. Increasing photoelectrocatalytic
k1 values of 6.80 × 10
−5 s
−1 (
R2 = 0.998), 1.09 × 10
−4 s
−1 (
R2 = 0.995), 1.42 × 10
−4 s
−1 (
R2 = 0.995), 1.57 × 10
−4 s
−1 (
R2 = 0.998), 1.68 × 10
−4 s
−1 (
R2 = 0.997), and 1.70 × 10
−4 s
−1 (
R2 = 0.995) were determined for E
cell at 0, 4.0, 8.0, 16.0, 32.0, and 62.0 V, respectively. Applying a small E
cell difference of 4.0 V resulted in a 1.6-fold increase in
k1 compared with the pure photocatalytic treatment. Further increases in E
cell provided only minor improvements in acetaminophen abatement. Thus, a maximum separation of charge carriers was attained at a certain applied E
cell, and further increases from this optimum potential did not improve performance.
The above changes in E
cell correspond with E
EO values decreasing from 130 kWh m
−3 order
−1 without a current down to 83 kWh m
−3 order
−1 at 4.0 V and 67 kWh m
−3 order
−1 at 8.0 V. Stabilization of oxidant species on the photoanode surface favors reactions that degrade acetaminophen [
2,
48]. However, additional increase in E
cell reduces the energy efficiency of the system, skyrocketing the energy requirements by an order of magnitude to 330 kWh m
−3 order
−1 at 62.0 V. This high E
EO can be explained by the concomitant acceleration of parasitic reactions induced by the increase of potential and current circulated during the electrochemically-driven process [
49,
50]. The excess energy is then consumed on the evolution of oxygen from water oxidation according to Equation (8) and/or dimerization of
•OH from Equation (9). Effects on the electrode characteristics and its morphology were not observed.
3.4. Effect of Acetaminophen Concentration on Photoelectrocatalytic Degradation
Initial pollutant concentrations may affect photoelectrocatalytic treatment performance by competing for surface sites [
2,
12] or absorbing light in solution, which prevents activation of the semiconductor TiO
2 nanotubes.
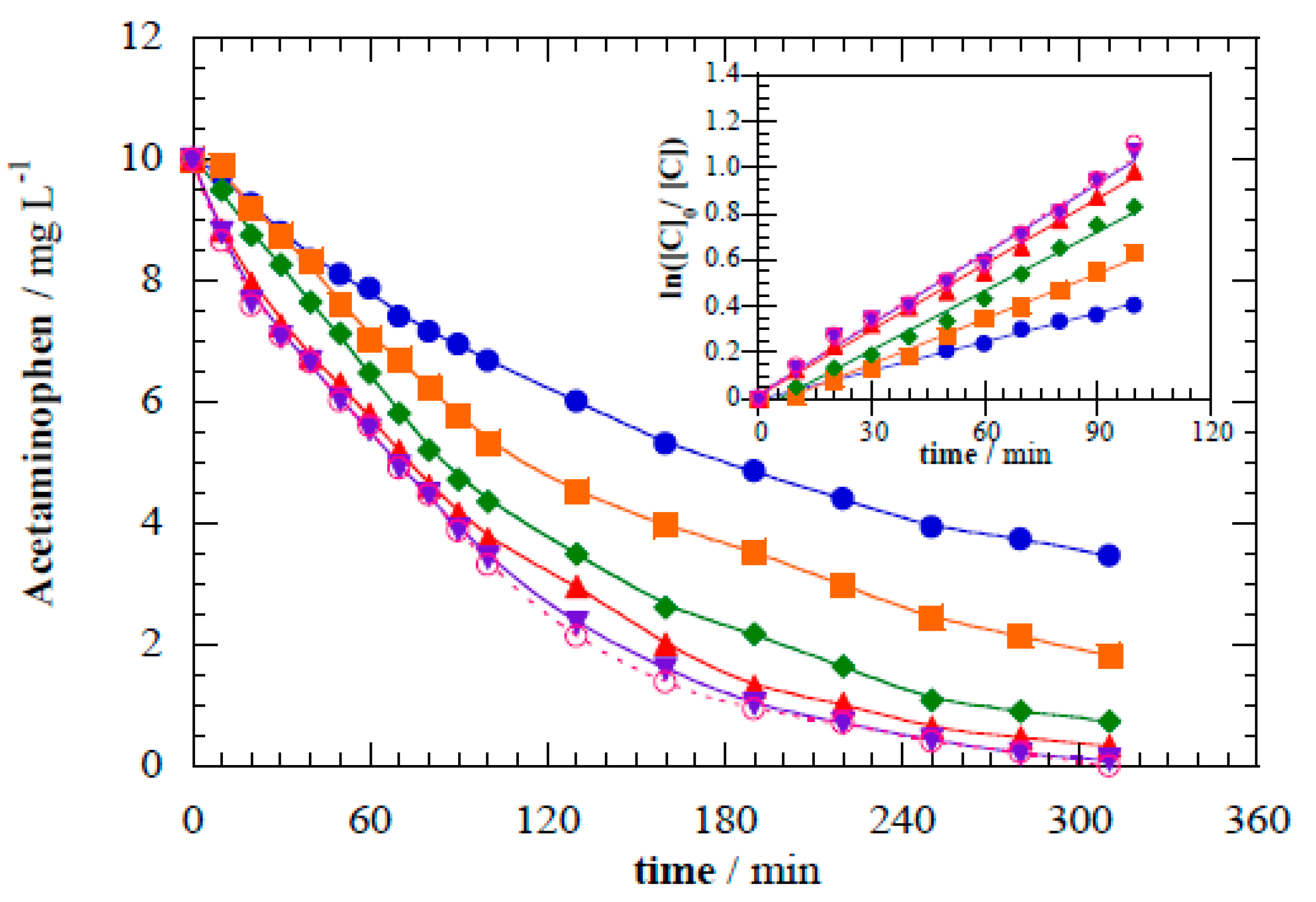
Figure 6 depicts the degradation kinetics for initial acetaminophen concentrations ranging from 5 mg L
−1 to 50 mg L
−1 in the photoelectrocatalytic operational mode. Kinetics analyses resulted in photoelectrocatalytic
k1 values of 2.05 × 10
−4 s
−1 for 5 mg L
−1 (
R2 = 0.996), 1.42 × 10
−4 s
−1 for 10 mg L
−1 (
R2 = 0.995), 6.66 × 10
−5 s
−1 for 25 mg L
−1 (
R2 = 0.999), 3.18 × 10
−5 s
−1 for 35 mg L
−1 (
R2 = 0.999), and 2.86 × 10
−5 s
−1 for 50 mg L
−1 (
R2 = 0.998).
Although acetaminophen has a low quantum yield (Φ
253 = 0.006), direct absorption of photons by acetaminophen in solution can explain the slower removal observed at higher pollutant concentration (
Figure 6) [
29,
39]. Absorption diminishes photon transport efficiency and limits the amount of light reaching the photoelectrode [
23,
29]. Photon flux is directly related to the efficient generation of charge carriers by Equation (1); consequently, reducing the photon delivery will also reduce the oxidants photogenerated [
6,
7]. Furthermore, a driving effect on the deceleration is the higher concentration of organics in solution that compete for h
vb+ and
•OH.
The decrease in photogeneration efficiency and the competition for surface sites increases the energy costs required for operation. As initial concentration increased, so did EEO for the photoelectrocatalytic treatment: 47 kWh m−3 order−1 for 5 mg L−1, 67 kWh m−3 order−1 for 10 mg L−1, 144 kWh m−3 order−1 for 25 mg L−1, 300 kWh m−3 order−1 for 30 mg L−1, and 334 kWh m−3 order−1 for 50 mg L−1. It may be inferred that photoelectrocatalytic treatment would be better suited as a polishing step to reduce the concentration of highly-persistent organic pollutants such as pharmaceuticals that may be found in fine chemicals, manufacturing effluents, or hospital effluents. However, photoelectrocatalysis should be conducted in water with high transmittance to allow UV light to reach the electrode surfaces.
3.5. Photoelectrocatalytic Mineralization and Time-Course of Yielded Byproducts.
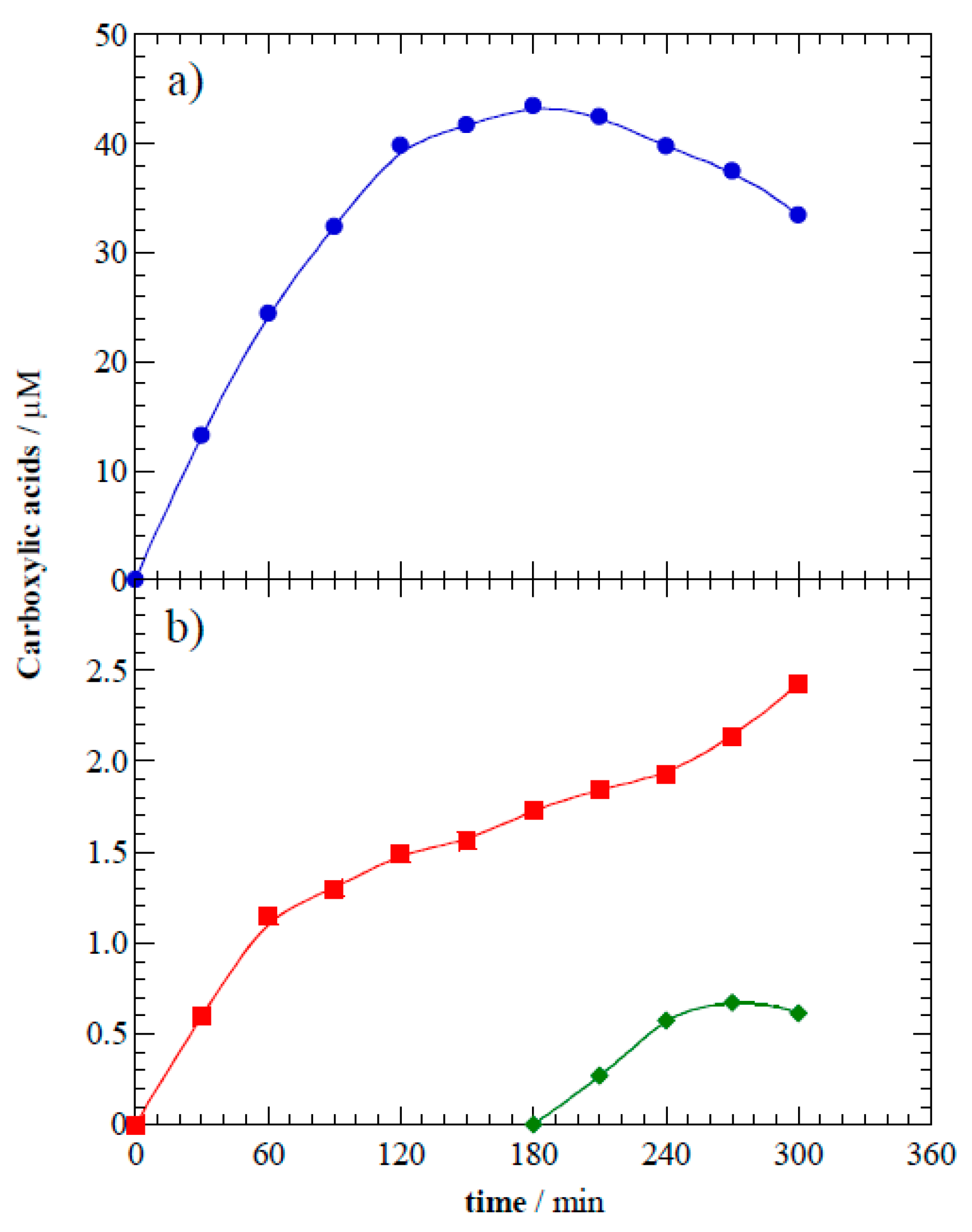
Acetaminophen was slowly mineralized during photoelectrocatalytic treatment.
Figure 7a shows the treated solution attained 53% mineralization at the point when >95% parent compound removal occurred. The remaining TOC suggests that organic by-products are accumulated. Oxidation reactions mediated by h
vb+ and
•OH lead to the aromatic moiety opening and yield short-linear aliphatic carboxylic acids [
2]. Ion-exclusion chromatography was used to measure formic, oxamic, and oxalic acids. These highly recalcitrant species are the ultimate products released during AOPs mediated by
•OH prior to the complete mineralization of organics to CO
2.
Figure 8 shows that these carboxylic acids accumulated in solution and reached final concentrations of 33.4 μM formic acid, 2.4 μM oxalic acid, and 0.6 μM oxamic acid. After their formation, the carboxylic acids remained in solution because they are very slowly degraded. This result agrees with previous reports that carboxylic acids are hardly oxidized by h
vb+ and
•OH but are biodegradable [
16,
40].
As a result of oxidizing the amide functional group, acetaminophen degradation is accompanied by inorganic nitrogen species formation (see
Figure 7b inset). Anionic chromatography identified NO
3− release at maximum concentration of 0.005 mM. Nitrite formation was not observed, which is a common trend of AOPs where the high oxidative media of ROS favors complete oxidation towards NO
3− [
51,
52]. Ammonium was continuously released from the breakage of the amide bond and reached a maximum concentration of 0.021 mM. To better understand the fate of the initial N-organic content, a complete mass balance was conducted (
Figure 7b). Compared with the 53% mineralization and volatilization of carbon, only 46% volatilization of nitrogen occurred as the likely result of producing volatile species such as N2 and NOx [
53,
54,
55]. The remaining nitrogen in solution was associated to remaining acetaminophen (0.005 mM), yielded oxamic acid (0.61 μM), and dissolved inorganic nitrogen (0.026 mM); inorganic nitrogen represented 75% of total dissolved nitrogen in solution after photoelectrocatalytic treatment.
4. Conclusions
A monopolar, multiple-disc composite photoelectrocatalytic reactor was constructed to overcome limitations of light transport in photoelectrocatalytic reactors. The parallel arrangement of disc photoanodes and cathodes defined a hydraulic pathway for the recirculated solution while providing the requirements for homogeneous current distribution in electrolytic reactors and light delivery for charge carriers’ photogeneration. This novel reactor system can provide an alternative framework when considering the scaling-up of photoelectrocatalytic water treatment.
Negligible acetaminophen removal was observed by direct photolysis or electrocatalysis. Photoelectrocatalytic treatment was more energy efficient than photocatalysis alone because applying a bias cell potential prevented recombination of ROS on photocatalytic nanotubes’ surfaces. Therefore, EEO was reduced two fold from 130 kWh m−3 order−1 for pure photocatalytic treatment down to 67 kWh m−3 order−1 for photoelectrocatalytic treatment.
Applying Ecell improved charge separation and stabilized ROS on the electrode surface. However, when more than 8.0 V was delivered, the electrical current was consumed in parasitic reactions, increasing EEO. Further increases in Ecell did not improve reactor performance and would have a detrimental impact operational cost.
Pollutant concentration diminished the light transport efficiency, which reduced photon delivery to the photoelectrode surface and reduced charge carrier generation. Even though high concentrations of acetaminophen may be treated in the photoelectrocatalytic reactor, EEO would increase due to a reduction in treatment performance.
These results demonstrated that scaling-up reactor designs for photoelectrocatalytic treatments must fulfill the needs of light-driven and electrochemically-driven processes to retain performance with respect to efficient energy usage and pollutant removal. The use of multiple, concentric, monopolar discs appears to be a feasible approach. This initial proof of concept provides opportunity for additional photoelectrocatalytic reactor design research. Future work must evaluate ways to improve light transport, which may have been limited by the perpendicular orientation of the electrodes in this design.
Author Contributions
Conceptualization: R.M.-A., S.C., and S.G.-S.; methodology: R.M.-A., S.C., J.C.M.-G., and S.G.-S.; validation: R.M.-A., H.A., and P.W.; formal analysis, J.C.M. and S.C.; investigation, R.M.-A. and J.C.M.-G.; resources: H.A., P.W. and S.G.-S; data curation: R.M.-A. and S.C.; writing—original draft preparation, R.M.-A. and S.G.-S.; writing—review and editing, P.W. and S.G.-S; visualization, P.W. and S.G.-S.; supervision, J.C.M.-G. and S.G.-S.; project administration, H.A. and S.G.-S.; funding acquisition, H.A., P.W. and S.G.-S.
Funding
This work was partially funded by the National Science Foundation (NSF) through the Nanotechnology-Enabled Water Treatment Nanosystems Engineering Research Center under project EEC-1449500 and the Research Institute of the Universidad de Lima and FONDECYT-Perú under project 105-2015.
Acknowledgments
Authors acknowledge the technical editing provided by Laurel Passantino.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Minakata, D.; Li, K.; Westerhoff, P.; Crittenden, J. Development of a group contribution method to predict aqueous phase hydroxyl radical (HO•) reaction rate constants. Environ. Sci. Technol. 2009, 43, 6220–6227. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Segura, S.; Brillas, E. Applied photoelectrocatalysis on the degradation of organic pollutants in wastewaters. J. Photochem. Photobiol. C Photochem. Rev. 2017, 31, 1–35. [Google Scholar] [CrossRef]
- Komtchou, S.; Dirany, A.; Drogui, P.; Delegan, N.; El Khakani, M.A.; Robert, D.; Lafrance, P. Degradation of atrazine in aqueous solution with electrophotocatalytic process using TiO2-x photoanode. Chemosphere 2016, 157, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Mais, L.; Mascia, M.; Palmas, S.; Vacca, A. Photoelectrochemical oxidation of phenol with nanostructured TiO2-PANI electrodes under solar light irradiation. Sep. Purif. Technol. 2019, 208, 153–159. [Google Scholar] [CrossRef]
- Loeb, S.K.; Alvarez, P.J.J.; Brame, J.A.; Cates, E.L.; Choi, W.; Crittenden, J.; Dionysiou, D.D.; Li, Q.; Li-puma, G.; Quan, X.; et al. The Technology Horizon for Photocatalytic Water Treatment: Sunrise or Sunset? Environ. Sci. Technol. 2019, 53, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Tugaoen, H.O.N.; Garcia-Segura, S.; Hristovski, K.; Westerhoff, P. Challenges in photocatalytic reduction of nitrate as a water treatment technology. Sci. Total Environ. 2017, 599–600, 1524–1551. [Google Scholar] [CrossRef]
- Fagan, R.; Mccormack, D.E.; Dionysiou, D.D.; Pillai, S.C. A review of solar and visible light active TiO2 photocatalysis for treating bacteria, cyanotoxins and contaminants of emerging concern. Mater. Sci. Semicond. Process. 2016, 42, 2–14. [Google Scholar] [CrossRef]
- Marcelino, R.B.P.; Amorim, C.C. Towards visible-light photocatalysis for environmental applications: Band-gap engineering versus photons absorption—A review. Environ. Sci. Pollut. Res. 2019, 26, 4155–4170. [Google Scholar] [CrossRef]
- Cardoso, J.C.; Bessegato, G.G.; Boldrin Zanoni, M.V. Efficiency comparison of ozonation, photolysis, photocatalysis and photoelectrocatalysis methods in real textile wastewater decolorization. Water Res. 2016, 98, 39–46. [Google Scholar] [CrossRef]
- Dosta, S.; Robotti, M.; Garcia-Segura, S.; Brillas, E.; Cano, I.G.; Guilemany, J.M. Influence of atmospheric plasma spraying on the solar photoelectro-catalytic properties of TiO2 coatings. Appl. Catal. B Environ. 2016, 189, 151–159. [Google Scholar] [CrossRef]
- Yang, T.; Xue, J.; Tan, H.; Xie, A.; Li, S.; Yan, W.; Shen, Y. Highly ordered ZnO/ZnFe2O4 inverse opals with binder-free heterojunction interfaces for high-performance photoelectrochemical water splitting. J. Mater. Chem. 2018, 6, 1210–1218. [Google Scholar] [CrossRef]
- Bessegato, G.G.; Cardoso, J.C.; Zanoni, M.V.B. Enhanced photoelectrocatalytic degradation of an acid dye with boron-doped TiO2 nanotube anodes. Catal. Today 2015, 240, 100–106. [Google Scholar] [CrossRef]
- Pablos, C.; Marugán, J.; Adán, C.; Osuna, M.; van Grieken, R. Performance of TiO2 photoanodes toward oxidation of methanol and E. coli inactivation in water in a scaled-up photoelectrocatalytic reactor. Electrochim. Acta 2017, 258, 599–606. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, Y.; Bi, Y. Spatial dual-electric fields for highly enhanced the solar water splitting of TiO2 nanotube arrays. Nano Energy 2019, 57, 542–548. [Google Scholar] [CrossRef]
- Li, Z.; Shi, L.; Franklin, D.; Koul, S.; Kushima, A.; Yang, Y. Drastic enhancement of photoelectrochemical water splitting performance over plasmonic Al@TiO2 heterostructured nanocavity arrays. Nano Energy 2018, 51, 400–407. [Google Scholar] [CrossRef]
- AlSalka, Y.; Granone, L.I.; Ramadan, W.; Hakki, A.; Dillert, R.; Bahnemann, D.W. Iron-based photocatalytic and photoelectrocatalytic nano-structures: Facts, perspectives, and expectations. Appl. Catal. B Environ. 2019, 244, 1065–1095. [Google Scholar] [CrossRef]
- Martins, A.S.; Cordeiro-Junior, P.J.M.; Bessegato, G.G.; Carneiro, J.F.; Zanoni, M.V.B.; Lanza, M.R. de V. Electrodeposition of WO3 on Ti substrate and the influence of interfacial oxide layer generated in situ: A photoelectrocatalytic degradation of propyl paraben. Appl. Surf. Sci. 2019, 464, 664–672. [Google Scholar] [CrossRef]
- Orimolade, B.O.; Koiki, B.A.; Peleyeju, G.M.; Arotiba, O.A. Visible light driven photoelectrocatalysis on a FTO/BiVO4/BiOI anode for water treatment involving emerging pharmaceutical pollutants. Electrochim. Acta 2019, 307, 285–292. [Google Scholar] [CrossRef]
- Bedoya-Lora, F.E.; Hankin, A.; Kelsall, G.H. En route to a unified model for photo-electrochemical reactor optimisation. i - Photocurrent and H2 yield predictions. J. Mater. Chem. A 2017, 5, 22683–22696. [Google Scholar] [CrossRef]
- Hankin, A.; Bedoya-Lora, F.E.; ONG, C.K.; Alexander, J.C.; Petter, F.; Kelsall, G.H. From millimetres to metres: The critical role of current density distributions in photo-electrochemical reactor design. Energy Environ. Sci. 2017, 10, 346–360. [Google Scholar] [CrossRef]
- Bu, Y.; Ao, J.P. A review on photoelectrochemical cathodic protection semiconductor thin films for metals. Green Energy Environ. 2017, 2, 331–362. [Google Scholar] [CrossRef]
- Phuan, Y.W.; Ong, W.J.; Chong, M.N.; Ocon, J.D. Prospects of electrochemically synthesized hematite photoanodes for photoelectrochemical water splitting: A review. J. Photochem. Photobiol. C Photochem. Rev. 2017, 33, 54–82. [Google Scholar] [CrossRef]
- Spasiano, D.; Marotta, R.; Malato, S.; Fernandez-Ibañez, P.; Di Somma, I. Solar photocatalysis: Materials, reactors, some commercial, and pre-industrialized applications. A comprehensive approach. Appl. Catal. B Environ. 2015, 170–171, 90–123. [Google Scholar] [CrossRef]
- Phuan, Y.W.; Ismail, H.M.; Garcia-Segura, S.; Chong, M.N. Design and CFD modelling of the anodic chamber of a continuous PhotoFuelCell reactor for water treatment. Process Saf. Environ. Prot. 2017, 111, 449–461. [Google Scholar] [CrossRef]
- Kim, S.D.; Cho, J.; Kim, I.S.; Vanderford, B.J.; Snyder, S.A. Occurrence and removal of pharmaceuticals and endocrine disruptors in South Korean surface, drinking, and waste waters. Water Res. 2007, 41, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Radjenović, J.; Petrović, M.; Barceló, D. Fate and distribution of pharmaceuticals in wastewater and sewage sludge of the conventional activated sludge (CAS) and advanced membrane bioreactor (MBR) treatment. Water Res. 2009, 43, 831–841. [Google Scholar] [CrossRef]
- Sengupta, A.; Lyons, J.M.; Smith, D.J.; Drewes, J.E.; Snyder, S.A.; Heil, A.; Maruya, K.A. The occurrence and fate of chemicals of emerging concern in coastal urban rivers receiving discharge of treated municipal wastewater effluent. Environ. Toxicol. Chem. 2014, 33, 350–358. [Google Scholar] [CrossRef]
- Díaz-Real, J.A.; Ortiz-Ortega, E.; Gurrola, M.P.; Ledesma-Garcia, J.; Arriaga, L.G. Light-harvesting Ni/TiO2 nanotubes as photo-electrocatalyst for alcohol oxidation in alkaline media. Electrochim. Acta 2016, 206, 388–399. [Google Scholar] [CrossRef]
- Garcia-Segura, S.; Tugaoen, H.O.N.; Hristovski, K.; Westerhoff, P. Photon flux influence on photoelectrochemical water treatment. Electrochem. Commun. 2018, 87, 63–65. [Google Scholar] [CrossRef]
- De Luna, M.D.G.; Laciste, M.T.; Tolosa, N.C.; Lu, M. Effect of catalyst calcination temperature in the visible light photocatalytic oxidation of gaseous formaldehyde by multi-element doped titanium dioxide. Environ. Sci. Pollut. Res. 2018, 25, 15216–15225. [Google Scholar] [CrossRef]
- Liang, K.; Tay, K.; Kupreeva, O.V.; Orekhovskaya, T.I.; Lazarouk, S.K.; Borisenko, V.E. Fabrication of double-walled titania nanotubes and their photocatalytic activity. ACS Sustain. Chem. Eng. 2014, 2, 991–995. [Google Scholar] [CrossRef]
- Hanaor, D.A.H.; Sorrell, C.C. Review of the anatase to rutile phase transformation. J. Mater. Sci. 2011, 48, 855–874. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent advances on developing 3rd generation enzyme electrode for biosensor applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Jianna, F.; Villaluz, A.; Daniel, M.; De Luna, G.; Colades, J.I.; Garcia-segura, S.; Lu, M. Removal of 4-chlorophenol by visible-light photocatalysis using ammonium iron (II) sulfate-doped nano-titania. Process Saf. Environ. Prot. 2019, 125, 121–128. [Google Scholar]
- Gualdrón-Reyes, A.F.; Meléndez, A.M.; González, I.; Lartundo-Rojas, L.; Niño-Gómez, M.E. Effect of Metal Substrate on Photo(electro)catalytic Activity of B-Doped Graphene Modified TiO2 Thin Films: Role of Iron Oxide Nanoparticles at Grain Boundaries of TiO2. J. Phys. Chem. C 2018, 122, 297–306. [Google Scholar] [CrossRef]
- Souza, F.L.; Aquino, J.M.; Miwa, D.W.; Rodrigo, M.A.; Motheo, A.J. Electrochemical degradation of dimethyl phthalate ester on a DSA® electrode. J. Braz. Chem. Soc. 2014, 25, 492–501. [Google Scholar] [CrossRef]
- Xiong, Y.; Yang, L.; Li, L.; Peng, L.; Liu, Y.; Luo, R.; Wei, X.; Xiao, P.; He, H. Interfacial engineering for the fast carrier tunneling channel by a novelty quaternary layered photoanode. ACS Appl. Energy Mater. 2019, 2, 6780–6789. [Google Scholar] [CrossRef]
- Cardoso, J.C.; Stulp, S.; de Brito, J.F.; Flor, J.B.S.; Frem, R.C.G.; Zanoni, M.V.B. MOFs based on ZIF-8 deposited on TiO2 nanotubes increase the surface adsorption of CO2 and its photoelectrocatalytic reduction to alcohols in aqueous media. Appl. Catal. B Environ. 2018, 225, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Fabrao, R.M.; de Brito, J.F.; da Silva, J.L.; Stradiotto, N.R.; Zanoni, M.V.B. Appraisal of photoelectrocatalytic oxidation of glucose and production of high value chemicals on nanotube Ti/TiO2 electrode. Electrochim. Acta 2016, 222, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Huitle, C.A.; Rodrigo, M.A.; Sirés, I.; Scialdone, O. Single and Coupled Electrochemical Processes and Reactors for the Abatement of Organic Water Pollutants: A Critical Review. Chem. Rev. 2015, 115, 13362–13407. [Google Scholar] [CrossRef]
- Cotillas, S.; De Vidales, M.J.M.; Llanos, J.; Sáez, C.; Cañizares, P.; Rodrigo, M.A. Electrolytic and electro-irradiated processes with diamond anodes for the oxidation of persistent pollutants and disinfection of urban treated wastewater. J. Hazard. Mater. 2016, 319, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.C.; Stefan, M.I.; Parnis, J.M.; Metcalfe, C.D. Direct UV photolysis of selected pharmaceuticals, personal care products and endocrine disruptors in aqueous solution. Water Res. 2015, 84, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, E.R.A.; Oliveira, G.A.R.; Grando, M.D.; Lizier, T.M.; Zanoni, M.V.B.; Oliveira, D.P. Photoelectrocatalysis based on Ti/TiO2 nanotubes removes toxic properties of the azo dyes Disperse Red 1, Disperse Red 13 and Disperse Orange 1 from aqueous chloride samples. J. Environ. Manage. 2013, 124, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Peleyeju, M.G.; Umukoro, E.H.; Tshwenya, L.; Moutloali, R.; Babalola, J.O.; Arotiba, O.A. Photoelectrocatalytic water treatment systems: Degradation, kinetics and intermediate products studies of sulfamethoxazole on a TiO2-exfoliated graphite electrode. RSC Adv. 2017, 7, 40571–40580. [Google Scholar] [CrossRef] [Green Version]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Cerrón-Calle, G.A.; Aranda-Aguirre, A.J.; Luyo, C.; Garcia-Segura, S.; Alarcón, H. Photoelectrocatalytic decolorization of azo dyes with nano-composite oxide layers of ZnO nanorods decorated with Ag nanoparticles. Chemosphere 2019, 219, 296–304. [Google Scholar] [CrossRef]
- Bessegato, G.G.; Cardoso, J.C.; da Silva, B.F.; Zanoni, M.V.B. Combination of photoelectrocatalysis and ozonation: A novel and powerful approach applied in Acid Yellow 1 mineralization. Appl. Catal. B Environ. 2016, 180, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Aquino, J.M.; Miwa, D.W.; Rodrigo, M.A.; Motheo, A.J. Treatment of actual effluents produced in the manufacturing of atrazine by a photo-electrolytic process. Chemosphere 2017, 172, 185–192. [Google Scholar] [CrossRef]
- Mostafa, E.; Reinsberg, P.; Garcia-Segura, S.; Baltruschat, H. Chlorine species evolution during electrochlorination on boron-doped diamond anodes: In-situ electrogeneration of Cl2, Cl2O and ClO2. Electrochim. Acta 2018, 281, 831–840. [Google Scholar] [CrossRef]
- Do Nascimento Brito, C.; Da Silva, D.R.; Garcia-Segura, S.; De Moura, D.C.; Martínez-Huitle, C.A. Indirect electrochemical oxidation of reactive blue 19 dye as a model organic substrate: Role of anode material and oxidants electrochemically generated. J. Electrochem. Soc. 2016, 163, E62–E69. [Google Scholar] [CrossRef]
- Garcia-Segura, S.; Lanzarini-Lopes, M.; Hristovski, K.; Westerhoff, P. Electrocatalytic reduction of nitrate: Fundamentals to full-scale water treatment applications. Appl. Catal. B Environ. 2018, 236, 546–568. [Google Scholar] [CrossRef]
- Garcia-Segura, S.; Mostafa, E.; Baltruschat, H. Could NOx be released during mineralization of pollutants containing nitrogen by hydroxyl radical? Ascertaining the release of N-volatile species. Appl. Catal. B Environ. 2017, 207, 376–384. [Google Scholar] [CrossRef]
- Heck, K.N.; Garcia-Segura, S.; Westerhoff, P.; Wong, M.S. Catalytic Converters for Water Treatment. Acc. Chem. Res. 2019, 52, 906–915. [Google Scholar] [CrossRef] [PubMed]
- De Vidales, M.J.M.; Millán, M.; Sáez, C.; Cañizares, P.; Rodrigo, M.A. What happens to inorganic nitrogen species during conductive diamond electrochemical oxidation of real wastewater? Electrochem. Commun. 2016, 67, 65–68. [Google Scholar] [CrossRef]
Figure 1.
(a) Photoelectrocatalytic reactor schematic, (b) photo-anode discs dimensions, (c) cathode discs dimensions, and (d) reactor sectional view.
Figure 1.
(a) Photoelectrocatalytic reactor schematic, (b) photo-anode discs dimensions, (c) cathode discs dimensions, and (d) reactor sectional view.
Figure 2.
Mercury lamp emission depicting a maximum emission at 253 nm (solid blue line), and molar absorptivity coefficient of acetaminophen at different wavelengths (dashed red line).
Figure 2.
Mercury lamp emission depicting a maximum emission at 253 nm (solid blue line), and molar absorptivity coefficient of acetaminophen at different wavelengths (dashed red line).
Figure 3.
(a) Scanning electron microscopy image of TiO2 nanotubes formed during electrochemical anodization. (b) Raman spectra depicting the characteristic anatase peaks formed after annealing TiO2 nanotubes at 450 °C. (c) X-ray diffractogram spectra indicating main peaks associated to anatase (A) and rutile (R) crystalline phases. (d) Linear sweep voltammetry of TiO2 nanotube electrode in 0.02 M of Na2SO4 and scan rate 100 mV s−1. Conditions: (1) in the dark in absence of acetaminophen, (2) in the dark with 10 mg L−1 of acetaminophen, and (3) under light irradiation at λ = 275 nm and with 10 mg L−1 of acetaminophen.
Figure 3.
(a) Scanning electron microscopy image of TiO2 nanotubes formed during electrochemical anodization. (b) Raman spectra depicting the characteristic anatase peaks formed after annealing TiO2 nanotubes at 450 °C. (c) X-ray diffractogram spectra indicating main peaks associated to anatase (A) and rutile (R) crystalline phases. (d) Linear sweep voltammetry of TiO2 nanotube electrode in 0.02 M of Na2SO4 and scan rate 100 mV s−1. Conditions: (1) in the dark in absence of acetaminophen, (2) in the dark with 10 mg L−1 of acetaminophen, and (3) under light irradiation at λ = 275 nm and with 10 mg L−1 of acetaminophen.
Figure 4.
Acetaminophen abatement during treatment of 10 mg L−1 of acetaminophen in 0.02 M Na2SO4 under different processes: (●) direct photolysis, (◆) photocatalysis with TiO2 nanotube discs, (■) electrocatalysis at 8.0 V of Ecell with TiO2 nanotube discs, and (▲) photoelectrocatalysis at 8.0 V of Ecell with TiO2 nanotube discs.
Figure 4.
Acetaminophen abatement during treatment of 10 mg L−1 of acetaminophen in 0.02 M Na2SO4 under different processes: (●) direct photolysis, (◆) photocatalysis with TiO2 nanotube discs, (■) electrocatalysis at 8.0 V of Ecell with TiO2 nanotube discs, and (▲) photoelectrocatalysis at 8.0 V of Ecell with TiO2 nanotube discs.
Figure 5.
Acetaminophen abatement during the treatment of 10 mg L−1 of acetaminophen in 0.02 M Na2SO4 during photoelectrocatalysis under different applied cell potentials: (●) 0 V (photocatalysis), (■) 4.0 V, (◆) 8.0 V, (▲) 16.0 V, (▼) 32.0 V, and (◯) 62.0 V. Inset panel shows the corresponding kinetic analysis considering a pseudo-first-order decay.
Figure 5.
Acetaminophen abatement during the treatment of 10 mg L−1 of acetaminophen in 0.02 M Na2SO4 during photoelectrocatalysis under different applied cell potentials: (●) 0 V (photocatalysis), (■) 4.0 V, (◆) 8.0 V, (▲) 16.0 V, (▼) 32.0 V, and (◯) 62.0 V. Inset panel shows the corresponding kinetic analysis considering a pseudo-first-order decay.
Figure 6.
Acetaminophen abatement during the photoelectrocatalytic treatment at cell potential 8.0 V for different initial concentrations of acetaminophen: (●) 5 mg L−1, (■) 10 mg L−1, (◆) 20 mg L−1, (▲) 35 mg L−1, and (▼) 50 mg L−1. Inset panel shows the corresponding kinetic analysis considering pseudo-first-order.
Figure 6.
Acetaminophen abatement during the photoelectrocatalytic treatment at cell potential 8.0 V for different initial concentrations of acetaminophen: (●) 5 mg L−1, (■) 10 mg L−1, (◆) 20 mg L−1, (▲) 35 mg L−1, and (▼) 50 mg L−1. Inset panel shows the corresponding kinetic analysis considering pseudo-first-order.
Figure 7.
(a) Total organic carbon abatement and (b) N-species evolution during the photoelectrocatalytic treatment at 8.0 V of 1 L of 10 mg L−1 of acetaminophen: (■) TOC, (•) total nitrogen, (▼) acetaminophen, (◆) oxamic acid (C2H3NO3), (▲) NO3−, and (△) NH4+.
Figure 7.
(a) Total organic carbon abatement and (b) N-species evolution during the photoelectrocatalytic treatment at 8.0 V of 1 L of 10 mg L−1 of acetaminophen: (■) TOC, (•) total nitrogen, (▼) acetaminophen, (◆) oxamic acid (C2H3NO3), (▲) NO3−, and (△) NH4+.
Figure 8.
Time course of carboxylic acids yielded as intermediates of photoelectrocatalytic degradation at 8.0 V of 10 mg L−1 of acetaminophen: (•) formic acid, (■) oxalic acid, and (◆) oxamic acid.
Figure 8.
Time course of carboxylic acids yielded as intermediates of photoelectrocatalytic degradation at 8.0 V of 10 mg L−1 of acetaminophen: (•) formic acid, (■) oxalic acid, and (◆) oxamic acid.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








